
Abstract
Zika virus (ZIKV) is an arbovirus belonging to the genus Flavivirus (family Flaviviridae) and was first described in 1947 in Uganda following blood analyses of sentinel Rhesus monkeys1. Until the twentieth century, the African and Asian lineages of the virus did not cause meaningful infections in humans. However, in 2007, vectored by Aedes aegypti mosquitoes, ZIKV caused the first noteworthy epidemic on the Yap Island in Micronesia2. Patients experienced fever, skin rash, arthralgia and conjunctivitis2. From 2013 to 2015, the Asian lineage of the virus caused further massive outbreaks in New Caledonia and French Polynesia. In 2013, ZIKV reached Brazil, later spreading to other countries in South and Central America3. In Brazil, the virus has been linked to congenital malformations, including microcephaly and other severe neurological diseases, such as Guillain–Barré syndrome4,5. Despite clinical evidence, direct experimental proof showing that the Brazilian ZIKV (ZIKVBR) strain causes birth defects remains absent6. Here we demonstrate that ZIKVBR infects fetuses, causing intrauterine growth restriction, including signs of microcephaly, in mice. Moreover, the virus infects human cortical progenitor cells, leading to an increase in cell death. We also report that the infection of human brain organoids results in a reduction of proliferative zones and disrupted cortical layers. These results indicate that ZIKVBR crosses the placenta and causes microcephaly by targeting cortical progenitor cells, inducing cell death by apoptosis and autophagy, and impairing neurodevelopment. Our data reinforce the growing body of evidence linking the ZIKVBR outbreak to the alarming number of cases of congenital brain malformations. Our model can be used to determine the efficiency of therapeutic approaches to counteracting the harmful impact of ZIKVBR in human neurodevelopment.
Similar content being viewed by others


Main
The recent increase in microcephaly cases in Brazil has been associated with the outbreak of Zika virus (ZIKV)7, originating from an Asian-lineage strain that can be spread by Aedes aegypti mosquitoes8. The Brazilian ZIKV (ZIKVBR) has been detected in the placenta and amniotic fluid of two women with microcephalic fetuses9,10,11 and in the blood of microcephalic newborns10,12, suggesting that the virus can cross the placental membrane. The virus has also been identified in the brains and retinas of microcephalic fetuses11,12,13. However, there is no direct evidence of the mechanism by which ZIKVBR causes brain malformations. A previous study revealed that the African ZIKV (ZIKVAF, strain MR-766) has the ability to infect human skin cells14. Neurons and astrocytes in the mouse brain could also be infected, inducing hippocampal degeneration and necrosis of pyriform cells 7 days post-infection15. More recently, ZIKVAF was also shown to infect human pluripotent stem cell (hPSC)-derived neural progenitor cells (NPCs) in vitro, which induced apoptotic cell death16. These studies were performed using the MR-766 ZIKVAF strain isolated in Uganda in 1947, which shares 87–90% sequence similarity with the Polynesian and Brazilian isolates3,17. Nevertheless, because severe congenital malformations were not reported for African isolates, there is a need to study the association of ZIKV with microcephaly and birth defects with isolates from affected localities, such as the ZIKVBR strain. Therefore, there is an urgent need to develop model systems to determine the relationship between infection with the ZIKVBR strain and birth defects.
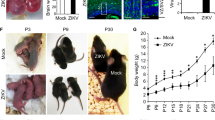
We used ZIKVBR isolated from a febrile case in the state of Paraiba, in the northeast of Brazil in 2015 (see Methods). To evaluate the causal relationship between ZIKVBR and birth defects, including brain malformation during development, we first used a murine experimental model in which SJL and C57BL/6 pregnant mice were infected with ZIKVBR, evaluating newborns immediately after birth (Extended Data Fig. 1a). Notably, similar to ZIKVBR-infected human newborns18,19, pups born from the SJL ZIKVBR-infected pregnant females displayed clear evidence of whole-body growth delay or intra-uterine growth restriction (IUGR)20 compared to pups born from the mock-infected controls (Fig. 1a, b). Using a qPCR assay, we confirmed the presence of ZIKVBR genomic RNA in several tissues of newborn animals, observing significantly more viral RNA in the brain, confirming the neurotropic nature of the virus (Fig. 1c).

a, SJL pups born with IUGR. Scale bar, 1 cm. b, Total body weight, crown–rump and skull measurements in pups born from infected animals (n = 6 pups, comprising 3 mice from 2 separate litters; error bars, s.e.m.; t-test, **P < 0.01). c, ZIKVBR RNA detected in SJL pup tissues (n = 6 pups, comprising 3 mice from 2 separate litters; error bars, s.e.m.; t-test). d, Histopathological aspect of the cortical organization (brackets) in infected brains, including intranuclear vacuoles, and ‘empty’ nuclei aspect with chromatin margination in neurons (arrowheads). Scale bar, 100 μm (left panels), 50 μm (middle panels) and 10 μm (right panels). e, ZIKVBR-infected brains displayed a reduced cortical layer thickness (n = 6 pups, comprising 3 mice from 2 separate litters; error bars, s.e.m.; t-test, ***P < 0.001). Infected brains have fewer cells per layer (n = 6 pups, comprising 3 mice from 2 separate litters; error bars, s.e.m.; t-test, **P < 0.1). f, ZIKVBR-infected cortical neurons have pronounced nuclei (diameter) (cortical, n = 31; deep cortical, n = 21 and medulla, n = 41 nuclei; error bars, s.e.m.; two-way ANOVA, ****P < 0.001). g, Ocular malformations (arrow) in the ZIKVBR-infected pups. h, Cell death gene expression signature in the brains of ZIKVBR-infected pups (n = 2 mice per group; threshold = twofold).
Microcephaly is perhaps the most dramatic of the birth defects reported in ZIKVBR-infected newborns4,18,19. Mouse models often fail to reproduce the severely reduced brain size and pathological alterations found in human patients21,22, probably owing to significant differences in gestation time and brain development between the two species. Nevertheless, upon close inspection of the ZIKVBR-infected mice brains, we noticed cortical malformations in the surviving animals, with reduced cell number and cortical layer thickness, signs associated with microcephaly in humans (Fig. 1d–f). At a cellular level, the neurons in the cortex, thalamus and hypothalamus displayed a ‘vacuolar nuclei’ appearance. This morphology was characterized by central emptiness and marginalized chromatin pattern with nuclear debris, suggesting ongoing cellular death (Fig. 1 d and Extended Data Fig. 2). In addition, we also noticed apparent ocular abnormalities, reminiscent of that observed in human patients23 (Fig. 1g). Thus, SJL infected pups presented congenital malformations compatible with ZIKVBR-infected human newborns. While the impact of ZIKVBR infection in the SJL mice was notable, no major body alterations were detected in pups from the infected C57BL/6 animals (Extended Data Fig. 1a–c). To exclude potential minor alterations in the C57BL/6 mice, computed tomography (CT) scans were performed to quantify the skull/body volumes. No significant changes in the pups born from the ZIKVBR-infected C57BL/6 mothers were observed compared to the controls (Extended Data Fig. 1d, e). A diagnostic qPCR assay of the pups from the ZIKVBR-infected animals was negative, suggesting that the virus did not cross the placenta in the C57BL/6 mouse strain (Extended Data Fig. 1f). To elucidate the type of cell death induced by ZIKVBR in the brains of SJL pups, we used a qPCR array to distinguish different molecular pathways involved. Our data clearly indicate that ZIKVBR infection influence the regulation of genes intimately linked to autophagy and apoptosis, such as upregulation of Bmf, Irgm1, Bcl2, Htt, Casp6 and Abl1. Conversely, Gadd45a, Tnfrsf11b, Fasl, Atg12, Bcl2l11 and Dffa were highly suppressed (Fig. 1h and Extended Data Fig. 1g, h).
Next, we evaluated the impact of ZIKVBR infection in human neural cells derived from hPSCs to establish a correlation between ZIKV and impairment of neurogenesis (Extended Data Fig. 3a). We generated human cortical NPCs and neurons from healthy donor hPSCs. First, we determined the expression levels of the TYRO3, AXL and MERTK (TAM) receptors tyrosine kinases in NPCs and neurons. This is an important family of receptors used for cell invasion by the Dengue virus and ZIKV, and AXL has been recently proposed as a candidate receptor for ZIKV infection during neurogenesis14,24. Mock-infected NPCs expressed higher levels of AXL when compared to mock-infected neurons (Fig. 2a). However, no significant changes in expression levels were observed upon ZIKV infection in NPCs (Fig. 2b). We then investigated the impact of ZIKVBR and ZIKVAF infection in NPCs and neurons. After infection using a viral multiplicity of infection (MOI) of 10, ZIKVBR particles were detected inside the NPCs and neurons at several stages of viral assembly using transmission electron microscopy (Fig. 2c). Immunostaining performed on NPCs and neurons at both an MOI of 10 and an MOI of 1 revealed production of viral protein aggregates (Fig. 2d and Extended Data Fig. 3b, c). With an MOI of 10, the amount of ZIKVBR particles in the NPC and neuron culture supernatant increased over time, suggesting the efficient production of infectious viral particles (Fig. 2e, f). With an MOI of 1, NPCs, but not neurons, continued to produce ZIKVBR RNA in the culture supernatant (Extended Data Fig. 3d, e). After 96 h post-infection we observed significant cell death in NPC cultures using fluorescence-activated cell (FAC) analyses. We quantified cell death over time in NPC cultures and detected an increase in the number of apoptotic/necrotic cells both in the ZIKVBR- and ZIKVAF-treated cultures compared to the mock-infected cultures at an MOI of 10 (Fig. 2g), but not at an MOI of 1 during the same time frame (Extended Data Fig. 3f). No difference in neuronal cell death was observed between the two ZIKV strains at an MOI of 10 and an MOI of 1 (Extended Data Fig. 3g, h).

a, Relative expression of TAM receptors (n = 2 technical replicates from two pooled different donors; error bars, s.e.m.; t-test; ***P < 0.01). b, Expression of TAM receptors in NPCs after ZIKVBR infection (MOI = 10) at 48 h post-infection (n = 2 technical replicates from two pooled different donors; error bars, s.e.m.; one-way ANOVA, P > 0.05). c, TEM detection of ZIKVBR viral particles 24 h post-infection at MOI = 10 (red arrowheads) inside NPCs (top panels) and neurons (bottom panels). Yellow arrowheads, viral factories; white arrowheads, immature viral particles. Scale bars/magnifications, 0.5 μm/40,000× (top left); 200 nm/ 80,000× (top right); 0.2 μm/ 50,000× (bottom left); 50 nm/ 300,000× (bottom right). d, Immunofluorescence revealed susceptibility to infection in NPC and neurons with the ZIKVBR (MOI = 10) at 24 h post-infection. Scale bar, 25 μm. e, ZIKVBR replication dynamics in NPCs (MOI = 10; n = 2 technical replicates from RNA of two different donors). f, ZIKVBR replication dynamics in neurons (MOI = 10; n = 2 technical replicates from RNA of two different donors). g, NPC death measured by FACS with two different cell gating sizes (P1 and P2). Apoptosis (left panel), necrosis (middle panel) and late apoptosis (right panel) (MOI = 10; 48 h post-infection; n = 2 technical replicates from two different donors; error bars, s.e.m.; two-way ANOVA, *P < 0.05). PI, propidium iodide. h, Representative images of human neurospheres infected with ZIKVBR (MOI = 10; 96 h post-infection). Scale bar, 200 μm. i, Alterations in neurosphere diameter over time (MOI = 10; n = 25 neurospheres from two different donors for each time point; error bars, s.e.m.; ANOVA, ****P < 0.0001). j, ZIKV replication dynamics in neurospheres (MOI = 10; n = 2 technical replicates from two different donors).
Next, we challenged two three-dimensional neural cell culture systems, neurospheres and cerebral organoids, with ZIKVBR and ZIKVAF. We generated neurospheres by growing human NPCs in suspension. While the mock-infected control neurospheres continued to grow over time, the ZIKVBR-infected neurospheres (MOI of 10) displayed evident morphological abnormalities with signs of cell death (Fig. 2h). The sizes of the neurospheres infected with ZIKVBR were significantly smaller than the mock-control and ZIKVAF-infected at 96 h post-infection (Fig. 2i). A less dramatic effect is observed at an MOI of 1, where both ZIKVBR and ZIKVAF infection reduced the size of the neurospheres compared to mock-infected controls (Extended Data Fig. 4a, b). These observations were paired with increased ZIKVBR replication in these cultures at both MOIs of 10 and 1 (Fig. 2j and Extended Data Fig. 4c). These results suggest that ZIKVBR induces cell death in human NPCs, impairing the growth and morphogenesis of healthy neurospheres (Extended Data Fig. 4d–f).
The majority of the described cases of ZIKVBR-infected newborns (95%) had malformations of cortical development4. Thus, we also used brain organoids generated from hPSCs and human embryonic stem cells, to evaluate the impact of ZIKVBR on human cortical development. In the following experiments, alongside the ZIKVAF and mock infection, we added the Yellow Fever virus (YFV), a slow replicating attenuated live-vaccine Flavivirus that has a low risk of causing neuropathy. Cerebral organoids are three-dimensional, self-organized, stem-cell-derived models that recapitulate the first trimester of human neurodevelopment, including the molecular and cellular architecture reminiscent of the fetal cortex25. Organoids show some degree of lamination and resembled the human neocortex in terms of the spatial relationships of the progenitor populations, defined here as a proliferative ventricular zone, post-migratory neurons in cortical plate and a marginal zone (Fig. 3a–c). We infected organoids with ZIKVBR, ZIKVAF and YFV using an MOI of 0.1 and compared to mock-infected organoids at 24 and 96 h post-infection. We quantified the percentage of different subtypes of cortical neurons, TBR1-positive or CTIP2-positive cells (deep-layer V/VI) in the cortical plate, finding a significant reduction in their number and respective cortical plate thickness in the ZIKVBR-infected organoids compared to the others. A significant reduction in TBR1-positive cells was observed in the ZIKVBR-infected organoids at 96 h post-infection, while CTIP2-positive cells were significantly reduced in both ZIKVBR- and ZIKVAF-infected organoids at the same time point (Fig. 3d, e and Extended Data Fig. 5a–f). Consistent with the reduced population of cortical neurons, we observed a significant decrease in PAX6-positive cells (dorsal forebrain progenitor cells) following ZIKV infection (Fig. 3f and Extended Data Fig. 5d). Dividing cells in the ventricular zone, detected by the population of KI67- and SOX2-positive cells, were only significantly reduced in the ZIKVBR-infected organoids (Fig. 3g, h and Extended Data Fig. 5d). As observed in our other in vitro models, the number of apoptotic cells (cleaved caspase 3- and TUNEL-positive cells) was increased in organoids infected with ZIKVBR, possibly explaining the decrease in the NPC population (Fig. 3i and Extended Data Fig. 5g, h).

a, Representative image of a human cerebral organoid showing the marginal zone (MZ), cortical plate (CP) and ventricular zone (VZ), delineated by dotted white lines. Scale bar, 200 μm. b, Representative images of the CP stained for CTIP2, TBR1, MAP2 or TUJ1 (neurons). Scale bar, 50 μm. c, Representative images of the proliferative regions in the VZ stained for KI67, PAX6, and cleaved caspase-3 (CASP3). Scale bar, 50 μm. d, Percentage of TBR1-positive cells in relation to mock-infected controls (dotted line) (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; ANOVA, **P = 0.0025). e, Percentage of CTIP2-positive cells (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; ANOVA, ****P < 0.001; *P = 0.0430). f, Percentage of PAX6-positive cells (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; ANOVA, *P = 0.0221). g, Percentage of KI67-positive cells (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; ANOVA, ****P < 0.001). h, Percentage of SOX2-positive cells (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; ANOVA, ****P = 0.0003). i, Percentage of cleaved-caspase3-positive cells (CASP3) (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; ANOVA, ****P = 0.0002).
ZIKVAF was derived from a zoonotic agent associated with primates in Africa, whereas ZIKVBR is an isolate from a lineage adapted to human-to-human transmission for the past 70 years. As an entry point to establishing the potential mechanistic adaptive differences between ZIKVBR and ZIKVAF towards human cells, we also generated brain organoids from non-human primate (chimpanzee) pluripotent stem cells. We infected these chimpanzee’s cerebral organoids (MOI of 0.1) and measured the impact on cortical neurons at 24 and 96 h post-infection. ZIKVBR failed to reduce the percentage of either TBR1- or CTIP2-positive cells in non-human primates (Extended Data Fig. 5i, j). Consistently, the kinetics of infection were different between the two ZIKV isolates. While ZIKVBR did not replicate in the chimpanzee organoids, ZIKVAF seemed well adapted to these cells (Extended Data Fig. 5k).
To evaluate the causal relationship between ZIKV congenital infection and birth defects, we used a murine experimental model, in which pregnant SJL and C57BL/6 mice were infected with ZIKVBR. Notably, the SJL strain was susceptible to viral infection of fetal tissues, causing severe IUGR that resembled the affected Brazilian newborns, including signs of microcephaly, such as cortical malformations4. We also showed that ZIKVBR induced apoptosis and autophagy in the mouse neural tissue. This is in accordance with the literature, as it has been previously demonstrated that ZIKV induces and localizes in autophagic phagosomes14. To our knowledge, this is the first report showing a gene expression profile that correlates to cell death in the brains of microcephalic newborn ZIKVBR-infected mice, corroborating a causal relationship. It is unclear why the virus could not cross the placenta of C57BL/6 mice, but this result may be due to the robust anti-viral immune response of this mouse strain, which secretes significant levels of type I/II interferon, known to confer resistance to ZIKV14,26. These data suggest that genetic differences could explain in part why some ZIKV-infected pregnant women give birth to newborns without detectable congenital brain malformations27. Nonetheless, our murine model is a valuable tool for future pre-clinical studies, such as vaccine development. The presence of major cortical histological abnormalities in the pups with IUGR prompted us to use an hPSC model to study the impact of ZIKV on neurodevelopment. ZIKV infects cells at different stages of brain maturation leading to alterations in the cortical layer organization. While this manuscript was under review, two other papers revealed the impact of previously established ZIKV strains on human organoids, confirming our observations with ZIKVBR (refs 28,29). Finally, our data using a non-human primate organoids suggested that the ZIKVBR might have experienced adaptive changes in human cells. In fact, it has been demonstrated that the Asian lineage of ZIKV is undergoing codon usage adaptation towards biases observed in highly expressed human genes30. Our findings support the hypothesis that microcephaly is a distinctive feature of recent ZIKV Asian-lineage virus, which originated in the Pacific and is now spreading in South and Central America.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment
Viral culture and amplification
A lyophilized ZIKV isolate from a clinical case in Brazil (ZIKVBR), provided by the Evandro Chagas Institute in Belém, Pará, was reconstituted in 0.5 ml of sterile DEPC water. The African-lineage MR-766 (ZIKVAF), a reference strain isolated in Uganda in 1947 and the Yellow Fever vaccine strain (YFV-17D)31, both used here as controls, were provided by the Institute Pasteur in Dakar, Senegal. Aedes albopictus mosquito cells (C6/36 cells) were previously prepared to culture the three viruses. C6/36 cell culture was maintained using Leibovitz’s L-15 medium supplemented with 10% fetal bovine serum (FBS) (Gibco), 1% non-essential amino acids (Gibco), 1% sodium pyruvate (Gibco), 1% penicillin/streptomycin (Gibco), 0.05% of amphotericin B (Gibco) and kept at 27 °C in the absence of CO2. After reaching an approximately 70% confluent monolayer, 50 μl of each viral sample was inoculated into C6/36 with an hour of adsorption, with gentle shaking every 10 min to allow the homogeneous adsorption of the viruses. At the end of the adsorption period, 5 ml of the culture media was added, plus 2% FBS, 1% non-essential amino acids and 1% sodium pyruvate. The cultures were then incubated under the same adsorption conditions. In the first subculture (T1), the infected cells were less confluent compared to the control cells but had few noticeable morphological changes. On the fourth day after infection, the second subculture (T2) was made blindly by transferring 500 μl of the T1 supernatant, followed by the third subculture, which was collected on the eighth day after infection (T3). Pronounced cytopathic effects were perceived beginning at T2. The supernatants were harvested, titrated and T3 was used for the experimental inoculation.
Virus titration
Titration (in PFU ml−1) of each C6/36 subculture was obtained by plaque assay to determine the amount of infectious viral particles (PFU). The virus titration was performed in porcine kidney epithelial (PS) cells and in L15 medium with 5% FBS. Briefly, the virus titration was done using 200 μl of L15 medium (5% FBS, 1% penicillin/streptomycin, and 1% glutamine) in a 24-well plate. Then, a serial dilution of each virus stock from ZIKVBR, ZIKVAF and YFV-17D in L15 medium was performed, from 10−1 to 10−11.Then, 200 μl of each dilution was added in each 24-well plate. After this, 1 × 106 PS cells were seeded in each well of a 24-well plate for at least 3 h at 37 °C to allow virus adsorption and PS cell adherence. Later, each well was overlaid with complete carboxymethyl cellulose (CMC) medium (0.6% in L15 supplemented with 3% FBS). After 5 days of incubation at 37 °C, the plaque visualization was made using blue-black staining solution. The most appropriate viral dilution was estimated to determine the amount of infected cells visible (PFU ml−1). For ZIKVBR, the first C6/36 subculture had a titre of 6 × 108. The following subcultures had titres of 7.5 × 106 (T2) and 4 × 1012 (T3). All the subculture aliquots were stored in cryovials and maintained in liquid nitrogen or were distributed to the ZIKV São Paulo task force.
In vivo infection
Pregnant mice, 6–8 weeks of age, C57BL/6 or SJL (JAX), were infected intravenously with 200 μl of ZIKVBR-infected C6/36 cell supernatant containing 1 × 103, 4 × 1010 or 1 × 1012 PFU m−1 of virus on day 10–13 of gestation. The animals were observed daily. All the experiments were performed with the approval of the Institute of Biomedical Sciences Ethics Committee protocol number 05/2016.
Real-time PCR
RNA was extracted from each sample (cells, supernatant of cell culture or mouse tissue) using the QIAamp UltraSens Virus Kit (Qiagen) or TRIzol reagent (Invitrogen). All RNA pellets were resuspended in 30 μl of RNase-free distilled water, quantified using a NanoDrop spectrophotometer (NanoDrop Technologies) and stored at −80 °C. The set of primers/probes specific for ZIKV were synthesized by Sigma Life Science, with 5- FAM as the reporter dye for the probe. The set of primers/probes ZIKV 835, ZIKV 911c and ZIKV 860-FAM were previously described2. The real-time reaction was performed with 10 μl of each sample and 10 μl of the AgPath-IDTM One-Step RT–PCR reagents (Applied Biosystems). The amplification was done in an Applied Biosystems 7500 real-time PCR system, and involved activation at 45 °C for 15 min, 95 °C for 15 min followed by 40 amplification cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 30 s. The real-time data were analysed using SDS software from Applied Biosystems. For the detection and quantification of viral RNA, the real-time PCR of each sample was compared with threshold cycle (Ct) value with a ZIKV plasmid standard curve, which was obtained carrying out serial dilutions of a clone of the envelope gene of an isolate from the 2007 Yap Island outbreak (provided by the Institute Pasteur in Dakar, Senegal). For the detection and quantification of YFV RNA, a YFV-specific real-time assay was applied. The fold changes of gene expression were calculated in comparison to the values for the YFV controls31. The positive PCR control Ct value was used to normalize gene expression and determine fold changes during the 96 hours post-infection. The RPLA27 gene (60S ribosomal protein L27) was used as endogenous control for the PCR reactions30. For the TAM receptors detection, NPCs or neurons from two donors were or were not infected with ZIKVBR, at 10 MOI for 48 h and were submitted to standard TRIzol (Invitrogen) protocol for RNA extraction. Total RNA was quantified using a NanoDrop spectrophotometer and 2 μg was used for further cDNA synthesis using the superscript III reverse-transcription polimerase (Invitrogen) according to manufacturer’s protocol. qPCR was performed using Taqman probes (Extended Data Table 1) and the QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems). For normalization ACTB was used as a housekeeping gene.
Cell death pathway analysis
One microgram of total RNA from brains of 4 pups, 2 pooled mock and 2 pooled ZIKVBR-infected from SJL mothers were submitted to gene expression analysis for cell death target genes using the RT2 Profiler PCR Array Mouse Cell Death PathwayFinder (cat. no. PAMM-212ZA- Qiagen) according to the manufacturer’s protocols. qPCR was performed in the QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems). To evaluate gene expression, we established a fold change threshold of at least twofold up- or downregulation compared to mock infected samples. Statistical analysis was performed using the RT2 profiler RT–PCR array data analysis software v3.5.
NPCs, neurons, neurospheres and organoids
We used three human and two chimpanzee iPSC clones that were previously characterized in the Beltrão-Braga and Muotri laboratories32,33,34,35 plus H9 human embryonic stem cells (hESC) for all the experiments using pluripotent stem cells. All the cell lines tested negative for mycoplasma contamination. Briefly, high passages of iPSC/hESC colonies on feeder-free plates were maintained for 5 days with mTSeR media (Stem Cell Technologies). On the fifth day, the medium was changed to N2 media (DMEM/F12 medium supplemented with 1X N2 supplement (Invitrogen) and the dual SMAD inhibitors, 1 μM dorsomorphin (Tocris) and 1 μM SB431542 (Stemgent), for 48 h. Further, the colonies were detached from the plate and cultured in suspension as embryoid bodies (EBs) for 5 days at 90 r.p.m. in N2 media with the dual SMAD inhibitors. The EBs were plated on matrigel-coated plates with NBF media composed of the following: DMEM/F12 media supplemented with 0.5X N2, 0.5X B27 supplement (Gibco), 20 ng ml−1 of FGF2 and 1% penicillin/streptomycin. The emerged rosettes containing the NPCs were manually picked, dissociated and plated in a double-coated plate with poly-ornithine (10 μg ml−1, Sigma-Aldrich) and laminin (2.5 μg ml−1, Gibco). The NPC population was expanded using NBF media. The neuronal differentiation induction protocol consisted of treating the confluent NPC plate with 10 μM ROCK inhibitor for 48 h (Y-27632, Calbiochem) in the absence of FGF in the media, with regular media changes every 3 or 4 days. Neurons were considered completely differentiated and ready to experiments after 28 days. To produce neurospheres, NPC were scrapped from the plates and submitted to continuous shaking for 5–7 days at 90 r.p.m. in NBF media. Cerebral organoids were generated as previously described25,36. All experiments were performed with the approval of the Institute of Biomedical Sciences Ethics Committee protocol number 1001.
In vitro infection
NPCs, neurons, neurospheres and organoids were infected with ZIKVBR, ZIKVAF, YFV and mock (culture supernatant from uninfected C6/36 cells). NPCs were seeded in plates in 24-well plates and after 24 h viral samples were diluted to the desired MOI (0.1; 1 or 10) and added to the cells. For viral adsorption, cells in monolayer were incubated for 1 h at 4 °C with gentle agitation every 10 min. Next, the inoculum was removed and cells were washed once with PBS (USB Corporation). Culture medium was added to each well, and cells were incubated at 37 °C and 5% CO2 for the duration of the experiment. For neurospheres, NPCs were kept in constant shaking. For neuronal infection, NPCs were previously differentiated for 28 days and then neurons were infected with the desired MOI. For organoids, the number of cells available for infection was estimated to be 2.5 × 104 cells, as calculated by dividing the average surface area of a typical organoid by the average area of a typical cell (that is, a fibroblast). This calculus was used to estipulate the desired 0.1 MOI. For mock controls, the same volume of supernatant was added to each experiment, and the same procedures were followed.
Immunofluorescence
Cells were fixed using paraformaldehyde, 4% in PBS, for 15 min at room temperature. After washing, the cells were permeabilized with 0.1% Triton X-100 (Promega) diluted in PBS for 15 min. After blocking with 2% of BSA (Sigma-Aldrich) for 4 h, primary antibodies directed against the following were added: anti-ZIKV (polyclonal mouse, Institute Pasteur in Dakar, 1:80), anti-Flavivirus D1-4G2-4-15 (polyclonal mouse, Millipore, 1:100), 1:50, anti-MAP2 (chicken, Abcam ab5392, 1:200), anti-cleaved-caspase-3 (rabbit, Cell Signaling #9661, 1:400), anti-Sox2 (mouse, Abcam ab97959), anti-GFAP (rabbit, Abcam, 1:500) and anti-Mushashi1 (rabbit, Abcam, 1:1000) (Extended Data Table 2). The cells were incubated overnight at 4 °C. Secondary antibodies were added for a one-hour incubation at room temperature: anti-mouse Alexa Fluor 488, anti-chicken Alexa Fluor 647, anti-rat Alexa Fluor 555 and anti-rabbit Alexa Fluor 555 (Invitrogen). The nuclei were stained with DAPI (Invitrogen, 1:10,000) diluted in a PBS 1× solution for 5 min and mounted with DPX (Sigma). Images were acquired with Nikon Eclipse 80i. Analysis of data was performed using software NIS Elements 3.22 (Tokyo, Japan).
Cerebral organoids analyses
Human and chimp organoids were infected with an MOI of 0.1 and analysed after 24 and 96 h post-infection. Organoids were cryo-sectioned at 20 μm. Immunofluorescence was performed after blocking sections in a solution with 0.1% Triton and 3% BSA (Gemini) for 1 h at room temperature. The primary antibodies were diluted in a solution with 0.1% Triton and 3% BSA, and the sections were incubated with following antibodies: anti-ZIKV, anti-Flavivirus, anti-MAP2, anti-cleaved-caspase-3 and anti-Sox2, all mentioned above, and anti-PAX6 (rabbit, Covance PRB-278P, 1:100), anti-TBR1 (rabbit, Abcam ab31940, 1:300), CTIP2 (rat, Abcam ab18465, 1:100) and Ki67 (rabbit, Abcam ab15580, 1:100). The sections were blocked with 0.1% Triton (Sigma-Aldrich) and 3% BSA for 30 min at room temperature and the secondary antibodies previously diluted, the same mentioned above, were added. The nuclei were stained with DAPI, as mentioned above and slides were mounted with DPX (Sigma-Aldrich).
Transmission electron microscopy
Cell pellet was fixed using a 3% glutaraldehyde solution (Merck) at 4 °C for 2 h, rinsed in three changes of PBS for 1 h, and incubated for 16 h at 4 °C. The next day, post-fixation was performed with 1% of osmium tetroxide for 30 min at room temperature. Dehydration was carried out gradually with a series of ethanol concentrations: 70%, 95% and 100%. Sample was taken through two changes of propylene oxide and placed at a 1:1 ratio with embedding medium for 1 h in a rotary mixer followed by 100% embedding medium at room temperature for 24 h. Fresh embedding medium was placed overnight at 37 °C and polymerized in oven to 24 h. Ultrathin sections were cut and stained with uranyl acetate and lead citrate. The cells were visualized with a transmission electron microscope (JEOL, JEM 1011, Peabody, Massachusetts, USA). All experimental analyses were performed blinded to the treatment.
Flow cytometry
Cells were infected with an MOI of 10 or 1, prepared using supernatants from infected C6/36, and equal volume of mock. Cellular infection occurred at 4 °C for 1 h with cell homogenization every 10 min. After that, the cells were washed once and then maintained at 37 °C in CO2 incubators with medium, as described before. After 24, 48, 72 and 96 h post-infection the cells were harvested and then submitted to a staining protocol for annexin V and propidium iodide (PI) (BD Biosciences). The cells were washed twice with PBS and were harvested with 200 μl of trypsin 0.25% (LGC) for 10 min at 37 °C. Next, the cell suspensions were washed in DMEM with 10% of FBS and centrifuged for 5 min at 450g and 4 °C. The cells were then resuspended in 20 μl of annexin V binding buffer in 96-well round-bottom plates and with 1 μl of FITC–annexin V + 1 μl of PI and then incubated at room temperature for 15 min protected from light. After incubation period, the samples were added to 80 μl of binding buffer and acquired in the BD FACS Accuri C6 (BD Biosciences) flow cytometer.
Computed tomography
Mice were properly anaesthetized with isoflurane and immobilized on their right side on the bed with a piece of gauze and positioned with the whole body in the field of view (FOV). CT images were acquired using small animal imaging equipment (Triumph Trimodality Gamma Medica Ideas) with 45 kVp, 0.4 mA and 2.13 min of X-ray exposition (512 projections over 360°). The images were reconstructed using the FBP (filtered back projection) algorithm, a matrix of 512 × 512, smoothing filter and a pixel size of 92–117 μm (according to the animal size). Experienced evaluators, who were blinded to the animal group assignments, performed images analyses using the AMIDE 1.0.4, General Public License software. Fiducial marks were added to measure the distance between points considering, visually, the larger axis of the brain in the sagittal plane for posterior–anterior and superior–inferior distances and the coronal plane for the lateral right–left. For measuring whole-body length, the distance between the superior point of the brain to the first vertebra of the tail was used. The thorax measure was made from the third rib (right–left) in the coronal plane to the spinal cord level. The results are presented in mm.
Histologic processing
Tissue histology was performed using a dehydrating protocol with two alcohol baths of 95% (the first for 1 h and 15 min and the second for 30 min), three absolute alcohol baths (1 h for the first followed by 3 h for the second and 2 h for the third), followed by clarification with three baths of xylene (the first for 30 min and the next two for 1 h each). Finally, the material was added to three paraffin baths (the first for 30 min and the second two for 1 h each). Then the material was immersed in paraffin and cut with a microtome to a thickness of 5 mm. The deparaffinization protocol consisted of three xylol baths heated in an oven for 30 min each, two baths of absolute alcohol for 2 min each, two baths of 95% alcohol lasting 2 min each, an alcohol in water bath (85%) for 2 min and the last bath in 70% alcohol for 2 min. The haematoxylin and eosin staining protocol began with two quick baths in running water, followed by a 2-min bath in distilled water, a 2-min bath in haematoxylin, a 5-min bath in running water, a 1-min bath in eosin, followed by 1-min in a fast-flowing water bath, two baths of 95% ethanol for 2 min each, two baths of absolute ethanol for 2 min each, ending with three baths in xylene for 2 min each. Slides were mounted using Permount (Sigma-Aldrich) and analysed on multiple coronal slices in glass slides using light microscopy (Olympus BX40, ZEISS KS400) on a genotype-blinded fashion.
References
Dick, G. W., Kitchen, S. F. & Haddow, A. J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 46, 509–520 (1952)
Lanciotti, R. S. et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 14, 1232–1239 (2008)
Faria, N. R. et al. Zika virus in the Americas: early epidemiological and genetic findings. Science 352, 345–349 (2016)
de Fatima Vasco Aragao, M. et al. Clinical features and neuroimaging (CT and MRI) findings in presumed Zika virus related congenital infection and microcephaly: retrospective case series study. Br. Med. J. 353, i1901 (2016)
Cao-Lormeau, V. M. et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539 (2016)
Rasmussen, S. A., Jamieson, D. J., Honein, M. A. & Petersen, L. R. Zika virus and birth defects - reviewing the evidence for causality. N. Engl. J. Med. http://dx.doi.org/10.1056/NEJMsr1604338 (2016)
Campos, G. S., Bandeira, A. C. & Sardi, S. I. Zika virus outbreak, Bahia, Brazil. Emerg. Infect. Dis. 21, 1885–1886 (2015)
Hayes, E. B. Zika virus outside Africa. Emerg. Infect. Dis. 15, 1347–1350 (2009)
Sarno, M. et al. Zika Virus infection and stillbirths: a case of hydrops fetalis, hydranencephaly and fetal demise. PLoS Negl. Trop. Dis. 10, e0004517 (2016)
Calvet, G. et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect. Dis. http://dx.doi.org/10.1016/S1473-3099(16)00095-5 (2016)
Martines, R. B. et al. Notes from the field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses - Brazil, 2015. MMWR Morb. Mortal. Wkly. Rep. 65, 159–160 (2016)
Mlakar, J. et al. Zika virus associated with microcephaly. N. Engl. J. Med. 374, 951–958 (2016)
Ventura, C. V., Maia, M., Bravo-Filho, V., Góis, A. L. & Belfort, R., Jr. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet 387, 228 (2016)
Hamel, R. et al. Biology of Zika virus infection in human skin cells. J. Virol. 89, 8880–8896 (2015)
Bell, T. M., Field, E. J. & Narang, H. K. Zika virus infection of the central nervous system of mice. Arch. Gesamte Virusforsch. 35, 183–193 (1971)
Tang, H. et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell http://dx.doi.org/10.1016/j.stem.2016.02.016 (2016)
Faye, O. et al. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop. Dis. 8, e2636 (2014)
Brasil, P. et al. Zika virus outbreak in Rio de Janeiro, Brazil: clinical characterization, epidemiological and virological aspects. PLoS Negl. Trop. Dis. 10, e0004636 (2016)
Brasil, P. et al. Zika virus infection in pregnant women in Rio de Janeiro - preliminary report. N. Engl. J. Med. http://dx.doi.org/10.1056/NEJMoa1602412 (2016)
Jang, E. A., Longo, L. D. & Goyal, R. Antenatal maternal hypoxia: criterion for fetal growth restriction in rodents. Front. Physiol. 6, 176 (2015)
Lizarraga, S. B. et al. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137, 1907–1917 (2010)
Pulvers, J. N. et al. Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc. Natl Acad. Sci. USA 107, 16595–16600 (2010)
de Paula Freitas, B. et al. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil. JAMA Ophthalmol. http://dx.doi.org/10.1001/jamaophthalmol.2016.0267 (2016)
Nowakowski, T. J. et al. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell http://dx.doi.org/10.1016/j.stem.2016.03.012 (2016)
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013)
Rossi, S. L. et al. Characterization of a novel murine model to study Zika virus. Am. J. Trop. Med. Hyg. http://dx.doi.org/10.4269/ajtmh.16-0111 (2016)
Cauchemez, S. et al. Association between Zika virus and microcephaly in French Polynesia, 2013-15: a retrospective study. Lancet http://dx.doi.org/10.1016/S0140-6736(16)00651-6 (2016)
Garcez, P. P. et al. Zika virus impairs growth in human neurospheres and brain organoids. Science http://dx.doi.org/10.1126/science.aaf6116 (2016)
Qian, X. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1–17 (2016)
Freire, C. C. d. M. et al. Spread of the pandemic Zika virus lineage is associated with NS1 codon usage adaptation in humans. BioRxiv http://biorxiv.org/content/early/2015/11/25/032839
Stock, N. K. et al. Biological and phylogenetic characteristics of yellow fever virus lineages from West Africa. J. Virol. 87, 2895–2907 (2013)
de Jonge, H. J. et al. Evidence based selection of housekeeping genes. PLoS One 2, e898 (2007)
Marchetto, M. C. et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539 (2010)
Marchetto, M. C. et al. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature 503, 525–529 (2013)
Beltrao-Braga, P. I. et al. Feeder-free derivation of induced pluripotent stem cells from human immature dental pulp stem cells. Cell Transplant. http://dx.doi.org/10.3727/096368911X566235 (2011)
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014)
Acknowledgements
This work was supported by grants from the Zika Network FAPESP projects 2011/18703-2 and 2014/17766-9, the NGO ‘The Tooth Fairy Project’, the National Institutes of Health through the U19MH107367, R01MH094753 and an NARSAD Independent Investigator Grant to A.R.M. We would like to thank M. Gonçalves and M. A. Miglino for scientific discussions, G. Amarante-Mendes for the FITC-Annexin V reagent, M. Zatz for the SJL mice, E. Durigon and his group for the ZIKVBR aliquots, P. Vasconcelos for providing a lyophilized ZIKVBR seed, N. Olsen for reagent and equipment support, D. Candido for analysis support, F. Mastrantonio for drawings and R. E. Grassi for electron and confocal microscopy support.
Author information
Authors and Affiliations
Contributions
F.R.G. derived the NPCs, neurons and neurospheres, performed the viral infections and cell analyses and analysed the data. I.R.F. performed the viral infections of cells, processed and analysed infected brain organoids, prepared manuscript figures and analysed the data. F.B.R. derived the NPCs, performed immunostainings and analyses, prepared manuscript figures and analysed the data. B.C.F. revised the manuscript and with S.R. generated the organoid cultures from iPSCs and assisted with the immunofluorescence staining and experimental design. J.L.M.D. performed macroscopic and microscopic staining and analyses of the mice. K.O.P.G. performed the TEM experiments, RNA extraction and quantification and histopathological analyses. C.B. and N.S. performed RNA extraction and quantification and prepared figures. G.C.P. performed cell cultures, analysed the data and revised the manuscript. C.M.P., I.C., C.L.F., W.N.B. and C.R. performed cell death qPCR assays and flow cytometry, D.G.A performed flow cytometry staining protocols and analysed the data. C.M.P., I.C. and D.G.A. infected and observed the pregnant mice daily. C.M.P., C.L.F., I.C. and C.R. processed the mouse tissues for virus quantification and further analyses. D.P.F., A.T.G. and C.A.B. performed the CT scans and analysed and plotted the data. C.T.B. and E.A.M. performed virus amplification, titration and gene expression analysis. A.A.S. provided MR766 and YFV-17D isolates and serological reagents. P.M.A.Z. designed the experiments and revised the manuscript. J.P.S.P., A.R.M. and P.C.B.B.-B. designed the experiments, analysed the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Impact of ZIKVBR infection in the C57BL/6 and SJL mice.
a, Scheme for infecting mice and the follow-up analyses. Pregnant females at approximately day 10–13 of gestation were challenged with 4 × 1010 PFU of ZIKVBR via an intravenous route. Their pups were analysed immediately after birth for signs of malformation. b, A representative pup from mock-infected and the ZIKVBR-infected C57BL/6 mice. Scale bar, 1 cm. c, C57BL/6 pups born with no gross morphological changes or size differences compared to mock controls (n = 21 pups from three separate litters, error bars, s.e.m, t-test). d, e, CT analysis confirmed lack of anatomical alterations (n = 21 pups from three separate litters, error bars, s.e.m, t-test). For scale, the crosses indicate 1.2 mm (top left; top right; bottom left) and 0.6 mm (bottom right). f, ZIKVBR RNA was not detected in the brains of six C57BL/6 pups. g, Cell death pathway signature revealed by qPCR gene expression in the brains of the ZIKVBR-infected SJL pups (n = 2 technical replicates of pooled RNA from two pups each group; threshold = twofold). h, Heat map representation of misregulated genes in g.
Extended Data Figure 2 Histopathological analysis of brains from ZIKVBR-infected SJL pups.
Morphological aspect of hippocampus, thalamus, hypothalamus and cerebellum from brains of pups born from mothers infected with ZIKVBR. Arrowheads indicate intranuclear vacuoles and ‘empty’ nuclei aspect with chromatin margination observed in thalamus and hypothalamus. Scale bar from left to right: 100 μm, 100 μm, 50 μm and 10 μm.
Extended Data Figure 3 Impact of ZIKV infection in human NPCs and neurons.
a, Scheme of the in vitro experiments using hPSCs. The cells were differentiated into NPCs, neurons, neurospheres and cerebral organoids to test the impact of ZIKVBR over time. b, Infection of NPCs with the ZIKVBR and ZIKVAF (MOI = 1) at 24 and 96 h post-infection. Scale bar, 25 μm. c, Aspects of iPSC-derived human neurons after ZIKV infection (MOI = 1) at 24 and 96 h post-infection. Scale bars, 200 μm (bright field); 25 μm (immunofluorescence). d, Viral replication dynamics in human NPCs over time (MOI = 1) (n = 2 technical replicates from two different donors; error bars, s.e.m.). e, Viral replication dynamics in human neurons over time (MOI = 1) (n = 2 technical replicates from two different donors; error bars, s.e.m.). f, Dynamics of NPCs toxicity over time after ZIKV infection (MOI = 1), indicating no significant differences among the different viruses (n = 2 technical replicates from two different donors; error bars, s.e.m.). g, h, Viral replication dynamics of ZIKV in human neurons over time at MOI = 10 and MOI = 1, respectively (n = 2 technical replicates from two different donors; error bars, s.e.m.; one-way ANOVA).
Extended Data Figure 4 Impact of ZIKV infection in human neurospheres.
a, Representative bright-field images of ZIKV infection (MOI = 1) at 24 and 96 h post-infection. Scale bar, 400 μm. b, Alterations in neurospheres diameter over time (MOI = 1; n = 22 neurospheres from two different donors for each time point in each condition; error bars, s.e.m.; ANOVA, *P = 0.0124 and ****P < 0.0001). c, ZIKV replication dynamics in neurospheres (MOI = 1; n = 3 technical replicates from two different donors). d, Representative bright-field images of ZIKV infection (MOI = 1) at 96 h post-infection. Scale bars, 400 μm and 1,000 μm (mock). The dotted circle describes the neurospheres borders indicating the immunostained regions in e and f. e, f, Immunostaining of neurospheres infected with ZIKV at 96 h post-infection at MOI = 1 (e) or MOI = 10 (f), revealing a qualitative reduction of proliferative cell migration from Mushashi (Mus)-positive cells. Scale bars, 50 μm.
Extended Data Figure 5 Human and chimp cerebral organoids infected with ZIKV.
a, Representative image of an entire cross-section of a cerebral human organoid infected with the ZIKVBR (MOI = 0.1, 24 h post-infection). Scale bar, 200 μm. b, Detail of the surface of a human organoid infected with the ZIKVBR at 24 h post-infection (MOI = 0.1). Marginal zone (MZ) and cortical plate (CP) delineated by dotted white lines. Scale bar, 200 μm. c, Detail of the surface of a human organoid infected with the ZIKVBR at 96 h post-infection (MOI = 0.1). Notice the significant tissue damage and reduction in the CP at 24 h post-infection. Scale bar, 200 μm. d, A representative characterization of CP and ventricular zone (VZ) in human organoid infected with the ZIKVBR at 24 and 96 h post-infection (MOI = 0.1). Scale bar, 50 μm. e, Reduction in the cortical thickness measured by the extension of TBR1-positive layer of cells in human organoids at 96 h post-infection (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; unpaired t-test, *P = 0.0203). f, Reduction in the cortical thickness measured by the extension of CTIP2-positive cells layer in human organoids at 96 h post-infection (MOI = 0.1; n = 3 replicates from three human cell lines; error bars, s.e.m.; unpaired t-test, ***P = 0.0005). g, Nuclear size (diameter) of cleaved-caspase-3-positive apoptotic cells in human organoids at 96 h post-infection (MOI = 0.1; n = 10 organoids/slides from three human cell lines; error bars, s.e.m.; unpaired t-test, ***P = 0.0004). Scale bar, 50 μm. h, Percentage of TUNEL-positive cells in relation to controls (dotted line) at 24 and 96 h post-infection (MOI = 0.1; n = 10 organoids/slides from three human cell lines; error bars, s.e.m.; ANOVA, **P = 0.0042). i, Percentage of TBR1-positive cells in non-primate organoids (chimp) in relation to controls (dotted line) at 24 and 96 h post-infection (MOI = 0.1; n = 3 organoids from two donors, error bars, s.e.m.; ANOVA). j, Percentage of CTIP2-positive cells in non-primate organoids (chimp) in relation to controls (dotted line) at 24 and 96 h post-infection (MOI = 0.1; n = 3 organoids from two donors; error bars, s.e.m.; ANOVA). k, Viral replication dynamics in chimpanzee organoids over time (MOI = 0.1; n = 3 replicates from two donors; error bars, s.e.m.; ANOVA).
Rights and permissions
About this article

Cite this article
Cugola, F., Fernandes, I., Russo, F. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271 (2016). https://doi.org/10.1038/nature18296
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature18296
This article is cited by
-
Repurposing of Zika virus live-attenuated vaccine (ZIKV-LAV) strains as oncolytic viruses targeting human glioblastoma multiforme cells
Journal of Translational Medicine (2024)
-
Interactions Between Extracellular Vesicles and Autophagy in Neuroimmune Disorders
Neuroscience Bulletin (2024)
-
Human pluripotent stem cell (hPSC) and organoid models of autism: opportunities and limitations
Translational Psychiatry (2023)
-
Human-specific genetics: new tools to explore the molecular and cellular basis of human evolution
Nature Reviews Genetics (2023)
-
Human 3D brain organoids: steering the demolecularization of brain and neurological diseases
Cell Death Discovery (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
