
Abstract
Tandem repeat proteins, which are formed by repetition of modular units of protein sequence and structure, play important biological roles as macromolecular binding and scaffolding domains, enzymes, and building blocks for the assembly of fibrous materials1,2. The modular nature of repeat proteins enables the rapid construction and diversification of extended binding surfaces by duplication and recombination of simple building blocks3,4. The overall architecture of tandem repeat protein structures—which is dictated by the internal geometry and local packing of the repeat building blocks—is highly diverse, ranging from extended, super-helical folds that bind peptide, DNA, and RNA partners5,6,7,8,9, to closed and compact conformations with internal cavities suitable for small molecule binding and catalysis10. Here we report the development and validation of computational methods for de novo design of tandem repeat protein architectures driven purely by geometric criteria defining the inter-repeat geometry, without reference to the sequences and structures of existing repeat protein families. We have applied these methods to design a series of closed α-solenoid11 repeat structures (α-toroids) in which the inter-repeat packing geometry is constrained so as to juxtapose the amino (N) and carboxy (C) termini; several of these designed structures have been validated by X-ray crystallography. Unlike previous approaches to tandem repeat protein engineering12,13,14,15,16,17,18,19,20, our design procedure does not rely on template sequence or structural information taken from natural repeat proteins and hence can produce structures unlike those seen in nature. As an example, we have successfully designed and validated closed α-solenoid repeats with a left-handed helical architecture that—to our knowledge—is not yet present in the protein structure database21.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
Accession codes
Primary accessions
Protein Data Bank
Data deposits
Crystal structures determined in this study have been deposited in the RCSB Protein Data Bank under accession numbers 4YXX (dTor_6x35L), 4YY2 (dTor_3x33L_2-2a), 4YY5 (dTor_3x33L_2-2b), 4YXY (dTor_9x31L_sub), 4YXZ (dTor_9x31L), and 5BYO (dTor_12x31L).
References
Marcotte, E. M., Pellegrini, M., Yeates, T. O. & Eisenberg, D. A census of protein repeats. J. Mol. Biol. 293, 151–160 (1999)
Kajava, A. V. Tandem repeats in proteins: from sequence to structure. J. Struct. Biol. 179, 279–288 (2012)
Andrade, M. A., Perez-Iratxeta, C. & Ponting, C. P. Protein repeats: structures, functions, and evolution. J. Struct. Biol. 134, 117–131 (2001)
Grove, T. Z., Cortajarena, A. L. & Regan, L. Ligand binding by repeat proteins: natural and designed. Curr. Opin. Struct. Biol. 18, 507–515 (2008)
Wang, X., McLachlan, J., Zamore, P. D. & Hall, T. M. Modular recognition of RNA by a human pumilio-homology domain. Cell 110, 501–512 (2002)
Mak, A. N., Bradley, P., Cernadas, R. A., Bogdanove, A. J. & Stoddard, B. L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335, 716–719 (2012)
Deng, D. et al. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335, 720–723 (2012)
Barkan, A. et al. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 8, e1002910 (2012)
Reichen, C., Hansen, S. & Plückthun, A. Modular peptide binding: from a comparison of natural binders to designed armadillo repeat proteins. J. Struct. Biol. 185, 147–162 (2014)
Wierenga, R. K. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 492, 193–198 (2001)
Kobe, B. & Kajava, A. V. When protein folding is simplified to protein coiling: the continuum of solenoid protein structures. Trends Biochem. Sci. 25, 509–515 (2000)
Main, E. R., Xiong, Y., Cocco, M. J., D’Andrea, L. & Regan, L. Design of stable alpha-helical arrays from an idealized TPR motif. Structure 11, 497–508 (2003)
Binz, H. K. et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nature Biotechnol. 22, 575–582 (2004)
Parmeggiani, F. et al. Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J. Mol. Biol. 376, 1282–1304 (2008)
Urvoas, A. et al. Design, production and molecular structure of a new family of artificial alpha-helicoidal repeat proteins (αRep) based on thermostable HEAT-like repeats. J. Mol. Biol. 404, 307–327 (2010)
Boersma, Y. L. & Plückthun, A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr. Opin. Biotechnol. 22, 849–857 (2011)
Rämisch, S., Weininger, U., Martinsson, J., Akke, M. & André, I. Computational design of a leucine-rich repeat protein with a predefined geometry. Proc. Natl Acad. Sci. USA 111, 17875–17880 (2014)
Voet, A. R. et al. Computational design of a self-assembling symmetrical β-propeller protein. Proc. Natl Acad. Sci. USA 111, 15102–15107 (2014)
Park, K. et al. Control of repeat-protein curvature by computational protein design. Nature Struct. Mol. Biol. 22, 167–174 (2015)
Parmeggiani, F. et al. A general computational approach for repeat protein design. J. Mol. Biol. 427, 563–575 (2015)
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000)
Leaver-Fay, A. et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 (2011)
Koga, N. et al. Principles for designing ideal protein structures. Nature 491, 222–227 (2012)
Kajander, T., Cortajarena, A. L., Mochrie, S. & Regan, L. Structure and stability of designed TPR protein superhelices: unusual crystal packing and implications for natural TPR proteins. Acta Crystallogr. D 63, 800–811 (2007)
Jiménez-Menéndez, N. et al. Human mitochondrial mTERF wraps around DNA through a left-handed superhelical tandem repeat. Nature Struct. Mol. Biol. 17, 891–893 (2010)
Aurora, R. & Rose, G. D. Helix capping. Protein Sci. 7, 21–38 (1998)
Wintjens, R. T., Rooman, M. J. & Wodak, S. J. Automatic classification and analysis of alpha alpha-turn motifs in proteins. J. Mol. Biol. 255, 235–253 (1996)
Grove, T. Z., Regan, L. & Cortajarena, A. L. Nanostructured functional films from engineered repeat proteins. J. R. Soc. Interface 10, http://dx.doi.org/10.1098/rsif.2013.0051 (2013)
Lanci, C. J. et al. Computational design of a protein crystal. Proc. Natl Acad. Sci. USA 109, 7304–7309 (2012)
Abe, S. & Ueno, T. Design of protein crystals in the development of solid biomaterials. RSC Adv. 5, 21366–21375 (2015)
Simons, K. T., Kooperberg, C., Huang, E. & Baker, D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 (1997)
Dantas, G. et al. High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J. Mol. Biol. 366, 1209–1221 (2007)
Holm, L. & Sander, C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 (1995)
Sillitoe, I. et al. CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 43, D376–D381 (2015)
Fox, N. K., Brenner, S. E. & Chandonia, J. M. SCOPe: Structural Classification of Proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 42, D304–D309 (2014)
Cheng, H. et al. ECOD: an evolutionary classification of protein domains. PLOS Comput. Biol. 10, e1003926 (2014)
Mak, A. N., Lambert, A. R. & Stoddard, B. L. Folding, DNA recognition, and function of GIY-YIG endonucleases: crystal structures of R.Eco29kI. Structure 18, 1321–1331 (2010)
Walden, H. Selenium incorporation using recombinant techniques. Acta Crystallogr. D 66, 352–357 (2010)
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997)
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007)
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011)
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010)
Skubák, P., Murshudov, G. N. & Pannu, N. S. Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D 60, 2196–2201 (2004)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012)
He, J. et al. The structure of the 26S proteasome subunit Rpn2 reveals its PC repeat domain as a closed toroid of two concentric α-helical rings. Structure 20, 513–521 (2012)
Acknowledgements
The authors thank Scientific Computing at the Fred Hutchinson Cancer Research Center for providing the computational infrastructure necessary for this project. This research was supported by the following research grants: National Institutes of Health R21GM106117 to P.B. and R01GM49857 to B.L.S., and Swiss National Science Foundation Postdoc Fellowship PBZHP3-125470 and Human Frontier Science Program Long-Term Fellowship LT000070/2009-L to F.P.
Author information
Authors and Affiliations
Contributions
L.D., J.H., J.B. and F.P. expressed, purified, and characterized designed constructs. L.D., J.H. and J.B. performed crystal screening, collected diffraction data, and solved crystal structures. P.B. developed and implemented the repeat design algorithms. P.B. performed sequence design calculations with feedback from F.P. P.B., B.L.S. and D.B. supervised the research. P.B. conceived of the toroid design project with input from B.L.S. and D.B. P.B. wrote the manuscript with input from the other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
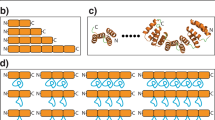
Extended Data Figure 1 Handedness of α-helical bundles and helical linkers.
a, Design dTor_12x31L, shown on the left, has a left-handed helical bundle. The native toroid on the right, which has a right-handed bundle, is taken from the Protein Data Bank structure 4ADY and corresponds to the PC repeat domain of the 26S proteasome subunit Rpn2 (ref. 46). b, The handedness of a helical bundle is determined by the twist direction of the polypeptide chain as it wraps around the axis of the helical bundle. c, Helical linkers characterized by a negative (positive) dihedral angle between the axes of the connected helices will, upon repetition, tend to impart a left-handed (right-handed) twist to the bundle. d, Geometrical properties of the most common short α-helical linkers in the structural database indicate that certain turn types (for example, ‘E’ and ‘GBB’) tend to form left-handed connections whereas others (for example, ‘GB’ and ‘BAAB’) are associated with right-handed connections. Turn types are classified by mapping their backbone torsion angles to a coarse-grained alphabet27 as shown in e.
Extended Data Figure 2 Unbiased 2Fo − Fc omit maps contoured around the side chains comprising the central pore regions for each crystallized toroid.
The constructs shown are in the same order as in Fig. 3.
Extended Data Figure 3 The crystallographic structures of highly symmetrical designed toroidal repeat proteins display rotational averaging in the crystal lattice.
a, Electron difference density for construct dTor_6x35L. Left: anomalous difference Fourier peaks calculated from data collected from a crystal of selenomethionine-derivatized protein. Although only one methionine residue (at position 168) is present in the construct, strong anomalous difference peaks (I/σI greater than 4.0) are observed at equivalent positions within at least three modular repeats. Right: difference density extending across the modelled position of the N and C termini in the refined model, indicating partial occupancy at that position by a peptide bond. The other five equivalent positions around the toroidal protein structure display equivalent features of density, indicating that each position is occupied by a mixture of loops and protein termini. b, Electron density for construct dTor_12x31L, again calculated at a position corresponding to the refined N and C termini in the crystallographic model. As was observed for the hexameric toroid in a, the electron density indicates a mixture of loops and protein termini.
Extended Data Figure 4 Size-exclusion chromatography elution profiles for the four designed toroids whose crystal structures were determined.
The elution profiles (blue traces) shown correspond to runs in high (750 mM) NaCl for dTor_3x33L_2-2 (a) and dTor_6x35L (b), while the elution profiles for dTor_9x31L (c) and dTor_12x31L (d) correspond to runs in lower (150 mM) NaCl. The superimposed elution profiles of standard protein size markers (brown traces) correspond to runs at those same salt concentrations, conducted on the same column and day. The inset in each panel displays the migration and relative purity of each construct used for the analysis.
Extended Data Figure 5 Purification and characterization of designed toroids.
a–g, CD wavelength scan from 260 to 190 nm of several designed toroids and a positive control protein at 22 °C (blue) and 80 °C (red). a, dTor_9x31L_sub; b, dTor_3x33L_2-2; c, dTor_6x33R_1; d, dTor_6x35L; e, dTor_9x31L; f, dTor_12x31L; g, positive control. h, Bis-Tris gel (4–12%) showing designed toroids immediately after metal affinity purification. Lane L, molecular mass protein standards (in kilodaltons); lane 1, dTor_9x31L_sub; lane 2, dTor_3x33L_2-2; lane 3, dTor_6x33R_1; lane 4, dTor_6x35L; lane 5, dTor_9x31L; lane 6, dTor_12x31L.
Extended Data Figure 6 Potential dimerization interfaces observed in crystal packing interactions.
a, Superposition of monomer–monomer packing interactions for the dTor_3x33L_2-2 design observed in two entirely different crystal forms. b, Stacking interactions between two dTor_6x35L subunits observed in the crystal structure; lysine residues interacting with backbone carbonyl groups in the partner monomer are shown in stick representation and coloured yellow along with their interaction partners.
Supplementary information
Supplementary information
This file contains a Supplementary Discussion, Supplementary Data and Supplementary References. (PDF 175 kb)
Rights and permissions
About this article

Cite this article
Doyle, L., Hallinan, J., Bolduc, J. et al. Rational design of α-helical tandem repeat proteins with closed architectures. Nature 528, 585–588 (2015). https://doi.org/10.1038/nature16191
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature16191
This article is cited by
-
Design of complicated all-α protein structures
Nature Structural & Molecular Biology (2024)
-
MPI-dot2dot: A parallel tool to find DNA tandem repeats on multicore clusters
The Journal of Supercomputing (2022)
-
De novo design of transmembrane nanopores
Science China Chemistry (2022)
-
Enumeration and comprehensive in-silico modeling of three-helix bundle structures composed of typical αα-hairpins
BMC Bioinformatics (2021)
-
Role of backbone strain in de novo design of complex α/β protein structures
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
