
Abstract
Coleoid cephalopods (octopus, squid and cuttlefish) are active, resourceful predators with a rich behavioural repertoire1. They have the largest nervous systems among the invertebrates2 and present other striking morphological innovations including camera-like eyes, prehensile arms, a highly derived early embryogenesis and a remarkably sophisticated adaptive colouration system1,3. To investigate the molecular bases of cephalopod brain and body innovations, we sequenced the genome and multiple transcriptomes of the California two-spot octopus, Octopus bimaculoides. We found no evidence for hypothesized whole-genome duplications in the octopus lineage4,5,6. The core developmental and neuronal gene repertoire of the octopus is broadly similar to that found across invertebrate bilaterians, except for massive expansions in two gene families previously thought to be uniquely enlarged in vertebrates: the protocadherins, which regulate neuronal development, and the C2H2 superfamily of zinc-finger transcription factors. Extensive messenger RNA editing generates transcript and protein diversity in genes involved in neural excitability, as previously described7, as well as in genes participating in a broad range of other cellular functions. We identified hundreds of cephalopod-specific genes, many of which showed elevated expression levels in such specialized structures as the skin, the suckers and the nervous system. Finally, we found evidence for large-scale genomic rearrangements that are closely associated with transposable element expansions. Our analysis suggests that substantial expansion of a handful of gene families, along with extensive remodelling of genome linkage and repetitive content, played a critical role in the evolution of cephalopod morphological innovations, including their large and complex nervous systems.
Similar content being viewed by others
Main
Soft-bodied cephalopods such as the octopus (Fig. 1a) show remarkable morphological departures from the basic molluscan body plan, including dexterous arms lined with hundreds of suckers that function as specialized tactile and chemosensory organs, and an elaborate chromatophore system under direct neural control that enables rapid changes in appearance1,8. The octopus nervous system is vastly modified in size and organization relative to other molluscs, comprising a circumesophageal brain, paired optic lobes and axial nerve cords in each arm2,3. Together these structures contain nearly half a billion neurons, more than six times the number in a mouse brain2,9. Extant coleoid cephalopods show extraordinarily sophisticated behaviours including complex problem solving, task-dependent conditional discrimination, observational learning and spectacular displays of camouflage1,10 (Supplementary Videos 1 and 2).

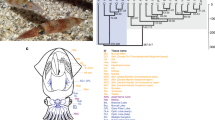
a, Schematic of Octopus bimaculoides anatomy, highlighting the tissues sampled for transcriptome analysis: viscera (heart, kidney and hepatopancreas), yellow; gonads (ova or testes), peach; retina, orange; optic lobe (OL), maroon; supraesophageal brain (Supra), bright pink; subesophageal brain (Sub), light pink; posterior salivary gland (PSG), purple; axial nerve cord (ANC), red; suckers, grey; skin, mottled brown; stage 15 (St15) embryo, aquamarine. Skin sampled for transcriptome analysis included the eyespot, shown in light blue. b, C2H2 and protocadherin domain-containing gene families are expanded in octopus. Enriched Pfam domains were identified in lophotrochozoans (green) and molluscs (yellow), including O. bimaculoides (light blue). For a domain to be labelled as expanded in a group, at least 50% of its associated gene families need a corrected P value of 0.01 against the outgroup average. Some Pfams (for example, Cadherin and Cadherin_2) may occur in the same gene, however multiple domains in a given gene were counted only once. Bfl, Branchiostoma floridae; Cel, Caenorhabditis elegans; Cgi, Crassostrea gigas; Cte, Capitella teleta; Dme, Drosophila melanogaster; Dre, Danio rerio; Gga, Gallus gallus; Hsa, Homo sapiens; Hro, Helobdella robusta; Lch, Latimeria chalumnae; Lgi, Lottia gigantea; Mmu, Mus musculus; Obi, O. bimaculoides; Pfu, Pinctada fucata; Xtr, Xenopus tropicalis.
To explore the genetic features of these highly specialized animals, we sequenced the Octopus bimaculoides genome by a whole-genome shotgun approach (Supplementary Note 1) and annotated it using extensive transcriptome sequence from 12 tissues (Methods and Supplementary Note 2). The genome assembly captures more than 97% of expressed protein-coding genes and 83% of the estimated 2.7 gigabase (Gb) genome size (Methods and Supplementary Notes 1, 2, 3). The unassembled fraction is dominated by high-copy repetitive sequences (Supplementary Note 1). Nearly 45% of the assembled genome is composed of repetitive elements, with two bursts of transposon activity occurring ∼25-million and ∼56-million years ago (Mya) (Supplementary Note 4).
We predicted 33,638 protein-coding genes (Methods and Supplementary Note 4) and found alternate splicing at 2,819 loci, but no locus showed an unusually high number of splice variants (Supplementary Note 4). A-to-G discrepancies between the assembled genome and transcriptome sequences provided evidence for extensive mRNA editing by adenosine deaminases acting on RNA (ADARs). Many candidate edits are enriched in neural tissues7 and are found in a range of gene families, including ‘housekeeping’ genes such as the tubulins, which suggests that RNA edits are more widespread than previously appreciated (Extended Data Fig. 1 and Supplementary Note 5).
Based primarily on chromosome number, several researchers proposed that whole-genome duplications were important in the evolution of the cephalopod body plan4,5,6, paralleling the role ascribed to the independent whole-genome duplication events that occurred early in vertebrate evolution11. Although this is an attractive framework for both gene family expansion and increased regulatory complexity across multiple genes, we found no evidence for it. The gene family expansions present in octopus are predominantly organized in clusters along the genome, rather than distributed in doubly conserved synteny as expected for a paleopolyploid12,13 (Supplementary Note 6.2). Although genes that regulate development are often retained in multiple copies after paleopolyploidy in other lineages, they are not generally expanded in octopus relative to limpet, oyster and other invertebrate bilaterians11,14 (Table 1 and Supplementary Notes 7.4 and 8).
Hox genes are commonly retained in multiple copies following whole-genome duplication15. In O. bimaculoides, however, we found only a single Hox complement, consistent with the single set of Hox transcripts identified in the bobtail squid Euprymna scolopes with PCR16. Remarkably, octopus Hox genes are not organized into clusters as in most other bilaterian genomes15, but are completely atomized (Extended Data Fig. 2 and Supplementary Note 9). Although we cannot rule out whole-genome duplication followed by considerable gene loss, the extent of loss needed to support this claim would far exceed that which has been observed in other paleopolyploid lineages, and it is more plausible that chromosome number in coleoids increased by chromosome fragmentation.
Mechanisms other than whole-genome duplications can drive genomic novelty, including expansion of existing gene families, evolution of novel genes, modification of gene regulatory networks, and reorganization of the genome through transposon activity. Within the O. bimaculoides genome, we found evidence for all of these mechanisms, including expansions in several gene families, a suite of octopus- and cephalopod-specific genes, and extensive genome shuffling.
In gene family content, domain architecture and exon–intron structure, the octopus genome broadly resembles that of the limpet Lottia gigantea17, the polychaete annelid Capitella teleta17 and the cephalochordate Branchiostoma floridae14 (Supplementary Note 7 and Extended Data Fig. 3). Relative to these invertebrate bilaterians, we found a fairly standard set of developmentally important transcription factors and signalling pathway genes, suggesting that the evolution of the cephalopod body plan did not require extreme expansions of these ‘toolkit’ genes (Table 1 and Supplementary Note 8.2). However, statistical analysis of protein domain distributions across animal genomes did identify several notable gene family expansions in octopus, including protocadherins, C2H2 zinc-finger proteins (C2H2 ZNFs), interleukin-17-like genes (IL17-like), G-protein-coupled receptors (GPCRs), chitinases and sialins (Figs 1b, 2 and 3; Extended Data Figs 4, 5, 6 and Supplementary Notes 8 and 10).

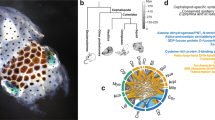
a, For a larger version of panel a, see Extended Data Fig. 11. Phylogenetic tree of cadherin genes in Hsa (red), Dme (orange), Nematostella vectensis (mustard yellow), Amphimedon queenslandica (yellow), Cte (green), Lgi (teal), Obi (blue), and Saccoglossus kowalevskii (purple). I, Type I classical cadherins; II, calsyntenins; III, octopus protocadherin expansion (168 genes); IV, human protocadherin expansion (58 genes); V, dachsous; VI, fat-like; VII, fat; VIII, CELSR; IX, Type II classical cadherins. Asterisk denotes a novel cadherin with over 80 extracellular cadherin domains found in Obi and Cte. b, Scaffold 30672 and Scaffold 9600 contain the two largest clusters of protocadherins, with 31 and 17, respectively. Clustered protocadherins vary greatly in genomic span and are oriented in a head-to-tail manner along each scaffold. c, Expression profiles of 161 protocadherins and 19 cadherins in 12 octopus tissues; 7 protocadherins were not detected in the tissues sampled. Cells are coloured according to number of standard deviations from the mean expression level. Protocadherins have high expression in neural tissues. Cadherins generally show a similar expression pattern, with the exception of a group of sucker-specific cadherins.

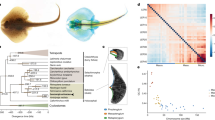
a, Genomic organization of the largest C2H2 cluster. Scaffold 19852 contains 58 C2H2 genes that are transcribed in different directions. b, Expression profile of C2H2 genes along Scaffold 19852 in 12 octopus transcriptomes. Neural and developmental transcriptomes show high levels of expression for a majority of these C2H2 genes. In a and b, arrow denotes scaffold orientation. c, Distribution of fourfold synonymous site transversion distances (4DTv) between C2H2-domain-containing genes.
The octopus genome encodes 168 multi-exonic protocadherin genes, nearly three-quarters of which are found in tandem clusters on the genome (Fig. 2b), a striking expansion relative to the 17–25 genes found in Lottia, Crassostrea gigas (oyster) and Capitella genomes. Protocadherins are homophilic cell adhesion molecules whose function has been primarily studied in mammals, where they are required for neuronal development and survival, as well as synaptic specificity18. Single protocadherin genes are found in the invertebrate deuterostomes Saccoglossus kowalevskii (acorn worm) and Strongylocentrotus purpuratus (sea urchin), indicating that their absence in Drosophila melanogaster and Caenorhabditis elegans is due to gene loss. Vertebrates also show a remarkable expansion of the protocadherin repertoire, which is generated by complex splicing from a clustered locus rather than tandem gene duplication (reviewed in ref. 19). Thus both octopuses and vertebrates have independently evolved a diverse array of protocadherin genes.
A search of available transcriptome data from the longfin inshore squid Doryteuthis (formerly, Loligo) pealeii20 also demonstrated an expanded number of protocadherin genes (Supplementary Note 8.3). Surprisingly, our phylogenetic analyses suggest that the squid and octopus protocadherin arrays arose independently. Unlinked octopus protocadherins appear to have expanded ∼135 Mya, after octopuses diverged from squid. In contrast, clustered octopus protocadherins are much more similar in sequence, either due to more recent duplications or gene conversion as found in clustered protocadherins in zebrafish and mammals21.
The expression of protocadherins in octopus neural tissues (Fig. 2) is consistent with a central role for these genes in establishing and maintaining cephalopod nervous system organization as they do in vertebrates. Protocadherin diversity provides a mechanism for regulating the short-range interactions needed for the assembly of local neural circuits18, which is where the greatest complexity in the cephalopod nervous system appears2. The importance of local neuropil interactions, rather than long-range connections, is probably due to the limits placed on axon density and connectivity by the absence of myelin, as thick axons are then required for rapid high-fidelity signal conduction over long distances. The sequence divergence between octopus and squid protocadherin expansions may reflect the notable differences between octopuses and decapodiforms in brain organization, which have been most clearly demonstrated for the vertical lobe, a key structure in cephalopod learning and memory circuits2,22. Finally, the independent expansions and nervous system enrichment of protocadherins in coleoid cephalopods and vertebrates offers a striking example of convergent evolution between these clades at the molecular level.
As with the protocadherins, we found multiple clusters of C2H2 ZNF transcription factor genes (Fig. 3a and Supplementary Note 8.4). The octopus genome contains nearly 1,800 multi-exonic C2H2-containing genes (Table 1), more than the 200–400 C2H2 ZNFs found in other lophotrochozoans and the 500–700 found in eutherian mammals, in which they form the second-largest gene family23. C2H2 ZNF transcription factors contain multiple C2H2 domains that, in combination, result in highly specific nucleic acid binding. The octopus C2H2 ZNFs typically contain 10–20 C2H2 domains but some have as many as 60 (Supplementary Note 8.4). The majority of the transcripts are expressed in embryonic and nervous tissues (Fig. 3b). This pattern of expression is consistent with roles for C2H2 ZNFs in cell fate determination, early development and transposon silencing, as demonstrated in genetic model systems23.
The expansion of the O. bimaculoides C2H2 ZNFs coincides with a burst of transposable element activity at ∼25 Mya (Fig. 3c). The flanking regions of these genes show a significant enrichment in a 70–90 base pair (bp) tandem repeat (31% for C2H2 genes versus 4% for all genes; Fisher’s exact test P value <1 × 10−16), which parallels the linkage of C2H2 gene expansions to β-satellite repeats in humans24. We also found an expanded C2H2 ZNF repertoire in amphioxus (Table 1), showing a similar enrichment in satellite-like repeats. These parallels suggest a common mode of expansion of a highly dynamic transcription factor family implicated in lineage-specific innovations.
To investigate further the evolution of gene families implicated in nervous system development and function, we surveyed genes associated with axon guidance (Table 1) and neurotransmission (Table 2), identifying their homologues in octopus and comparing numbers across a diverse set of animal genomes (Supplementary Notes 8, 9, 10). Several patterns emerged from this survey. The gene complements present in the model organisms D. melanogaster and C. elegans often showed striking departures from those seen in lophotrochozoans and vertebrates (Table 2 and Supplementary Note 10). For example, D. melanogaster encodes one member of the discs large (DLG) family, a key component of the postsynaptic scaffold. In contrast, mammals have four DLGs, which (along with other observations) led to suggestions that vertebrates possess uniquely complex synaptic machinery25. However, we found three DLGs in both octopus and limpet, suggesting that vertebrate and fly gene number differences are not necessarily diagnostic of exceptional vertebrate synaptic complexity (Supplementary Note 10.6).
Overall, neurotransmission gene family sizes in the octopus were very similar to those seen in other lophotrochozoans (Table 2 and Supplementary Note 10), except for a few strikingly expanded gene families such as the sialic acid vesicular transporters (sialins) (Supplementary Note 10.2). We did find variations in the sizes of neurotransmission gene families between human and lophotrochozoans (Table 2 and Supplementary Note 10), but no evidence for systematic expansion of these gene families in vertebrates relative to octopus or other lophotrochozoans. Although some gene families were larger in mammals or absent in lophotrochozoans (for example, ligand-gated 5-HT receptors), others were absent in mammals and present in invertebrates (for example, anionic glutamate and acetylcholine receptors). The complement of neurotransmission genes in octopus may be broadly typical for a lophotrochozoan, but our findings suggest it is also not obviously smaller than is found in mammals.
Among the octopus complement of ligand-gated ion channels, we identified a set of atypical nicotinic acetylcholine receptor-like genes, most of which are tandemly arrayed in clusters (Extended Data Fig. 7). These subunits lack several residues identified as necessary for the binding of acetylcholine26, so it is unlikely that they function as acetylcholine receptors. The high level of expression of these divergent subunits within the suckers raises the interesting possibility that they act as sensory receptors, as do some divergent glutamate receptors in other protostomes27. In addition, we identified 74 Aplysia-like and 11 vertebrate-like candidate chemoreceptors among the octopus GPCR superfamily of ∼330 genes (Extended Data Fig. 6).
We found, amid extensive transcription of octopus transposons, that a class of octopus-specific short interspersed nuclear element sequences (SINEs) is highly expressed in neural tissues (Supplementary Note 4 and Extended Data Fig. 8). Although the role of active transposons is unclear, elevated transposon expression in neural tissues has been suggested to serve an important function in learning and memory in mammals and flies28.
Transposable element insertions are often associated with genomic rearrangements29 and we found that the transposon-rich octopus genome displays substantial loss of ancestral bilaterian linkages that are conserved in other species (Supplementary Note 6 and Extended Data Fig. 9). Interestingly, genes that are linked in other bilaterians but not in octopus are enriched in neighbouring SINE content. SINE insertions around these genes date to the time of tandem C2H2 expansion (Extended Data Fig. 9d), pointing to a crucial period of genome evolution in octopus. Other transposons such as Mariner show no such enrichment, suggesting distinct roles for different classes of transposons in shaping genome structure (Extended Data Fig. 9c).
Transposable element activity has been implicated in the modification of gene regulation across several eukaryotic lineages29. We found that in the nervous system, the degree to which a gene’s expression is tissue-specific is positively correlated with the transposon load around that gene (r2 values ranging from 0.49 in the optic lobe to 0.81 in the subesophageal brain; Extended Data Fig. 8 and Supplementary Note 4). This correlation may reflect modulation of gene expression by transposon-derived enhancers or a greater tolerance for transposon insertion near genes with less complex patterns of tissue-specific gene regulation.
Using a relaxed molecular clock, we estimate that the octopus and squid lineages diverged ∼270 Mya, emphasizing the deep evolutionary history of coleoid cephalopods8,30 (Supplementary Note 7.1 and Extended Data Fig. 10a). Our analyses found hundreds of coleoid- and octopus-specific genes, many of which were expressed in tissues containing novel structures, including the chromatophore-laden skin, the suckers and the nervous system (Extended Data Fig. 10 and Supplementary Note 11). Taken together, these novel genes, the expansion of C2H2 ZNFs, genome rearrangements, and extensive transposable element activity yield a new landscape for both trans- and cis-regulatory elements in the octopus genome, resulting in changes in an otherwise ‘typical’ lophotrochozoan gene complement that contributed to the evolution of cephalopod neural complexity and morphological innovations.
Methods
Data access
Genome and transcriptome sequence reads are deposited in the SRA as BioProjects PRJNA270931 and PRJNA285380. The genome assembly and annotation are linked to the same BioProject ID. A browser of this genome assembly is available at (http://octopus.metazome.net/).
Genome sequencing and assembly
Genomic DNA from a single male Octopus bimaculoides31 was isolated and sequenced using Illumina technology to 60-fold redundant coverage in libraries spanning a range of pairs from ∼350 bp to 10 kilobases (kb). These data were assembled with meraculous32 achieving a contig N50-length of 5.4 kb and a scaffold N50-length of 470 kb. The longest scaffold contains 99 genes and half of all predicted genes are on scaffolds with 8 or more genes (Supplementary Note 1).
Genome size and heterozygosity
The O. bimaculoides haploid genome size was estimated to be ∼2.7 gigabases (Gb) based on fluorescence (2.66–2.68 Gb) and k-mer (2.86 Gb) measurements (Supplementary Notes 1 and 2), making it several times larger than other sequenced molluscan and lophotrochozoan genomes17. We observed nucleotide-level heterozygosity within the sequenced genome to be 0.08%, which may reflect a small effective population size relative to broadcast-spawning marine invertebrates.
Transcriptome sequencing
Twelve transcriptomes were sequenced from RNA isolated from ova, testes, viscera, posterior salivary gland (PSG), suckers, skin, developmental stage 15 (St15)33, retina, optic lobe (OL), supraesophageal brain (Supra), subesophageal brain (Sub), and axial nerve cord (ANC) (Supplementary Note 2). RNA was isolated using TRIzol (Invitrogen) and 100-bp paired-end reads (insert size 300 bp) were generated on an Illumina HiSeq2000 sequencing machine.
De novo transcriptome assembly
Adapters and low-quality reads were removed before assembling transcriptomes using the Trinity de novo assembly package (version r2013-02-25 (refs 34, 35)). Assembly statistics are summarized in Supplementary Table 2.2. Following assembly, peptide-coding regions were translated using TransDecoder in the Trinity package. We compared the de novo assembled RNA-seq output to the genome to evaluate the completeness of the genome assembly. To minimize the number of spuriously assembled transcripts, only transcripts with ORFs predicted by TransDecoder were mapped onto the genome with BLASTN. Only 1,130 out of 48,259 transcripts with ORFs (2.34%) did not have a match in the genome with a minimum identity of 95%.
Annotation of transposable elements
Transposable elements were identified with RepeatScout and RepeatModeler36, and the masking was done with RepeatMasker37, as outlined in Supplementary Note 4.2. The most abundant transposable element is a previously identified octopus-specific SINE38 that accounts for 4% of the assembled genome.
Annotation of protein-coding genes
Protein-coding genes were annotated by combining transcriptome evidence with homology-based and de novo gene prediction methods (Supplementary Note 4). For homology prediction we used predicted peptide sets of three previously sequenced molluscs (L. gigantea, C. gigas, and A. californica) along with selected other metazoans. Alternative splice isoforms were identified with PASA39. Annotation statistics are provided in Supplementary Table 4.1.1. Genes known in vertebrates to have many isoforms, such as ankyrin, TRAK1 and LRCH1, also show alternative splicing in octopus but at a more limited level. Octopus genes with ten or more alternative splice forms are provided in Supplementary Table 4.1.2.
Calibration of sequence divergence with respect to time
The divergence between squid and octopus was estimated using r8s40 by fixing cephalopod divergence from bivalves and gastropods to 540 Mya8. Our estimate of 270 Mya for the squid–octopus divergence corresponds to mean neutral substitution rate of dS ∼2 based on the protein-directed CDS alignments between the species (Supplementary Fig. 6.1.2) and a dS estimation using the yn00 program41. Throughout the manuscript we convert from sequence divergence to time by assuming that dS ∼1 corresponds to 135 million years. For example, unlinked octopus protocadherins appear to have expanded ∼135 Mya based on mean pairwise dS ∼1, after octopuses diverged from squid. In contrast, clustered octopus protocadherins are much more similar in sequence (mean pairwise dS ∼0.4, or ∼55 Mya).
Quantifying gene expression
Transcriptome reads were mapped to the genome assembly with TopHat 2.0.11 (ref. 42). A range of 76–90% of reads from the different samples mapped to the genome. Mapped reads were sorted and indexed with SAMtools43. The read counts in each tissue were produced with BEDTools multicov program44 using the gene model coordinates. The counts were normalized by the total transcriptome size of each tissue and by the length of the gene. Heat maps showing expression patterns were generated in R using the heatmap.2 function.
Gene complement
Gene families of particular interest, including developmental regulatory genes, neural-related genes, and gene families that appear to be expanded in O. bimaculoides, were manually curated and analysed. We searched the octopus genome and transcriptome assemblies using BLASTP and TBLASTN with annotated sequences from human, mouse and D. melanogaster. Bulk analyses were also performed using Pfam45 and PANTHER46. We used BLASTP and TBLASTX to search for specific gene families in deposited genome and transcriptome databases for L. gigantea, A. californica, C. gigas, C. teleta, T. castaneum, D. melanogaster, C. elegans, N. vectensis, A. queenslandica, S. kowalevskii, B. floridae, C. intestinalis, D. rerio, M. musculus and H. sapiens. Candidate genes were verified with BLAST47 and Pfam45 analysis. Genes identified in the octopus genome were confirmed and extended using the transcriptomes. Multiple gene models that matched the same transcript were combined. The identified sequences from octopus and other bilaterians were aligned with either MUSCLE48, CLUSTALO49, MacVector 12.6 (MacVector, North Carolina), or Jalview50. Phylogenetic trees were constructed with FastTree51 using the Jones–Taylor–Thornton model of amino acid evolution, and visualized with FigTree v1.3.1.
Synteny
Microsynteny was computed based on metazoan node gene families (Supplementary Note 7). We used Nmax 10 (maximum of 10 intervening genes) and Nmin 3 (minimum of three genes in a syntenic block) according to the pipeline described in ref. 17 (Supplementary Note 6). To simplify gene family assignments we limited our analyses to 4,033 gene families shared among human, amphioxus, Capitella, Helobdella, Octopus, Lottia, Crassostrea, Drosophila and Nematostella. We required ancestral bilaterian syntenic blocks to have a minimum of one species present in both ingroups, or in one ingroup and one outgroup. To examine the effect of fragmented genome assemblies, we simulated shorter assemblies by artificially fragmenting genomes to contain on average 5 genes per scaffold (Supplementary Note 6).
In comparison with other bilaterian genomes, we find that the octopus genome is substantially rearranged. In looking at microsyntenic linkages of genes with a maximum of 10 intervening genes, we found that octopus conserves only 34 out of 198 ancestral bilaterian microsyntenic blocks; the limpet Lottia and amphioxus retain more than twice as many such linkages (96 and 140, respectively). This difference remains significant after accounting for genes missed through orthology assignment as well as simulations of shorter scaffold sizes (Supplementary Note 6; Extended Data Fig. 9b). Scans for intra-genomic synteny, and doubly conserved synteny with Lottia, were performed as described in Supplementary Note 6.
Transposable elements and synteny dynamics
The 5 kb upstream and downstream regions of genes were surveyed for transposable element (TE) content. For genes with non-zero TE load, their assignment to either conserved or lost bilaterian synteny in octopus was done using the microsynteny calculation described above. The number of genes for each category and TE class were as follows: 484 genes for retained synteny and 1,193 genes in lost synteny for all TE classes; 440 and 1,107, respectively, for SINEs; and 116 and 290, respectively, for Mariner. Wilcoxon U-tests for the difference of TE load in linked versus non-linked genes were conducted in R.
To assess transposon activity we assigned transcriptome reads aligned to 5,496,558 annotated transposon loci using BEDTools44. Of these, 2,685,265 loci showed expression in at least one of the tissues.
RNA editing
RNA-seq reads were mapped to the genome with TopHat52, and SAMtools43 was used to identify SNPs between the genomic and the RNA sequences. To identify polymorphic positions in the genome, SNPs and indels were predicted using GATK HaplotypeCaller version 3.1-1 in discovery mode with a minimum Phred scaled probability score of 30, based on an alignment of the 350 bp and 500 bp genomic fragment libraries using BWA-MEM version 0.7.6a. Using BEDTools44, we removed SNPs predicted in both the transcriptome and the genome and discarded SNPs that had a Phred score below 40 or were outside of predicted genes. SNPs were binned according to the type of nucleotide change and the direction of transcription. Candidate edited genes were taken as those having SNPs with A-to-G substitutions in the predicted mRNA transcripts.
Cephalopod-specific genes
Cephalopod novelties were obtained by BLASTP and TBLASTN searches against the whole NR database53 and a custom database of several mollusc transcriptomes (Supplementary Note 11.1). To ensure that we had as close to full-length sequence as possible, we extended proteins predicted from octopus genomic sequence with our de novo assembled transcriptomes, using the longest match to query NR, transcriptome and EST sequences from other animals. Gene sequences with transcriptome support but without a match to non-cephalopod animals at an e-value cutoff of 1 × 10−3 were considered for further analysis. Octopus sequences with a match of 1 × 10−5 or better to a sequence from another cephalopod were used to construct gene families, which were characterized by their BLAST alignments, HMM, PFAM-A/B, and UNIREF90 hits. The cephalopod-specific gene families are listed in the Source Data file for Extended Data Fig. 10. Octopus-specific novelties were defined as sequences with transcriptome support but without any matches to sequences from any other animals (<1 × 10−3), including nautiloid and decapodiform cephalopods.
Accession codes
References
Hanlon, R. T. & Messenger, J. B. Cephalopod Behaviour (Cambridge Univ. Press, 1996)
Young, J. Z. The Anatomy of the Nervous System of Octopus vulgaris (Clarendon Press, 1971)
Wells, M. J. Octopus: Physiology and Behaviour of an Advanced Invertebrate (Chapman and Hall, 1978)
Bonnaud, L., Ozouf-Costaz, C. & Boucher-Rodoni, R. A molecular and karyological approach to the taxonomy of Nautilus. C. R. Biol. 327, 133–138 (2004)
Hallinan, N. M. & Lindberg, D. R. Comparative analysis of chromosome counts infers three paleopolyploidies in the mollusca. Genome Biol. Evol. 3, 1150–1163 (2011)
Yoshida, M. A. et al. Genome structure analysis of molluscs revealed whole genome duplication and lineage specific repeat variation. Gene 483, 63–71 (2011)
Rosenthal, J. J. & Seeburg, P. H. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 74, 432–439 (2012)
Kröger, B., Vinther, J. & Fuchs, D. Cephalopod origin and evolution. Bioessays 33, 602–613 (2011)
Herculano-Houzel, S., Mota, B. & Lent, R. Cellular scaling rules for rodent brains. Proc. Natl Acad. Sci. USA 103, 12138–12143 (2006)
Grasso, F. W. & Basil, J. A. The evolution of flexible behavioral repertoires in cephalopod molluscs. Brain Behav. Evol. 74, 231–245 (2009)
Holland, P. W., Garcia-Fernandez, J., Williams, N. A. & Sidow, A. Gene duplications and the origins of vertebrate development. Development (Suppl.), 125–133 (1994)
Dietrich, F. S. et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304, 304–307 (2004)
Kellis, M., Birren, B. W. & Lander, E. S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae . Nature 428, 617–624 (2004)
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071 (2008)
Duboule, D. The rise and fall of Hox gene clusters. Development 134, 2549–2560 (2007)
Callaerts, P. et al. HOX genes in the sepiolid squid Euprymna scolopes: implications for the evolution of complex body plans. Proc. Natl Acad. Sci. USA 99, 2088–2093 (2002)
Simakov, O. et al. Insights into bilaterian evolution from three spiralian genomes. Nature 493, 526–531 (2013)
Zipursky, S. L. & Sanes, J. R. Chemoaffinity revisited: Dscams, protocadherins, and neural circuit assembly. Cell 143, 343–353 (2010)
Chen, W. V. & Maniatis, T. Clustered protocadherins. Development 140, 3297–3302 (2013)
Brown, C. T., Graveley, B. & Rosenthal, J. J. Loligo pealeii (Squid) Data Dump (http://ivory.idyll.org/blog/2014-loligo-transcriptome-data.html) (2014)
Noonan, J. P., Grimwood, J., Schmutz, J., Dickson, M. & Myers, R. M. Gene conversion and the evolution of protocadherin gene cluster diversity. Genome Res. 14, 354–366 (2004)
Shomrat, T. et al. Alternative sites of synaptic plasticity in two homologous “fan-out fan-in” learning and memory networks. Curr. Biol. 21, 1773–1782 (2011)
Liu, H., Chang, L. H., Sun, Y., Lu, X. & Stubbs, L. Deep vertebrate roots for mammalian zinc finger transcription factor subfamilies. Genome Biol. Evol. 6, 510–525 (2014)
Eichler, E. E. et al. Complex β-satellite repeat structures and the expansion of the zinc finger gene cluster in 19p12. Genome Res. 8, 791–808 (1998)
Nithianantharajah, J. et al. Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nature Neurosci. 16, 16–24 (2013)
Brejc, K. et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411, 269–276 (2001)
Croset, V. et al. Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet. 6, e1001064 (2010)
Erwin, J. A., Marchetto, M. C. & Gage, F. H. Mobile DNA elements in the generation of diversity and complexity in the brain. Nature Rev. Neurosci. 15, 497–506 (2014)
Chénais, B., Caruso, A., Hiard, S. & Casse, N. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene 509, 7–15 (2012)
Strugnell, J., Norman, M., Jackson, J., Drummond, A. J. & Cooper, A. Molecular phylogeny of coleoid cephalopods (Mollusca: Cephalopoda) using a multigene approach; the effect of data partitioning on resolving phylogenies in a Bayesian framework. Mol. Phylogenet. Evol. 37, 426–441 (2005)
Pickford, G. E. & McConnaughey, B. H. The Octopus bimaculatus problem: a study in sibling species. Bulletin of the Bingham Oceanographic Collection 12, 1–66 (1949)
Chapman, J. A. et al. Meraculous: de novo genome assembly with short paired-end reads. PLoS ONE 6, e23501 (2011)
Naef, A., Boletzky, S. v. & Roper, C. F. E. Cephalopoda. Embryology (Smithsonian Institution Libraries, 2000)
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnol. 29, 644–652 (2011)
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512 (2013)
Smit, A. & Hubley, R. RepeatModeler Open-1.0. (–2008–2010)
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-3.0. (–1996–2010)
Ohshima, K. & Okada, N. Generality of the tRNA origin of short interspersed repetitive elements (SINEs). Characterization of three different tRNA-derived retroposons in the octopus. J. Mol. Biol. 243, 25–37 (1994)
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003)
Sanderson, M. J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302 (2003)
Yang, Z. & Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43 (2000)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25, 1105–1111 (2009)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010)
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014)
Mi, H., Muruganujan, A. & Thomas, P. D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 41, D377–D386 (2013)
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004)
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011)
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009)
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010)
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols 7, 562–578 (2012)
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005)
Palavicini, J. P., O'Connell, M. A. & Rosenthal, J. J. An extra double-stranded RNA binding domain confers high activity to a squid RNA editing enzyme. RNA 15, 1208–1218 (2009)
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001)
Starnes, T., Broxmeyer, H. E., Robertson, M. J. & Hromas, R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J. Immunol. 169, 642–646 (2002)
Cummins, S. F. et al. Candidate chemoreceptor subfamilies differentially expressed in the chemosensory organs of the mollusc Aplysia . BMC Biol. 7, 28 (2009)
van Nierop, P. et al. Identification of molluscan nicotinic acetylcholine receptor (nAChR) subunits involved in formation of cation- and anion-selective nAChRs. J. Neurosci. 25, 10617–10626 (2005)
Acknowledgements
We thank C. T. Brown and J. Rosenthal for making Doryteuthis RNA-seq data available before publication; C. Ha, J. Orenstein, J. Brandenburger, M. Glotzer and H. Gui for bioinformatic assistance; S. Shigeno for help with tissue dissection; C. Huffard and R. Caldwell for providing the O. bimaculoides specimen used for genomic DNA isolation; and E. Begovic for genomic DNA preparation. This work was supported by the Molecular Genetics Unit of the Okinawa Institute of Science and Technology Graduate University (S.B. and D.S.R.) and by funding from the NSF (IOS-1354898) and NIH (R03 HD064887) to C.W.R. and from the NSF (DGE-0903637) to Z.Y.W. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 instrumentation grants S10RR029668 and S10RR027303, and the University of Chicago Functional Genomics Facility, supported by NIH grant UL1 TR000430.
Author information
Authors and Affiliations
Contributions
The Chicago and the OIST/Berkeley groups initiated their transcriptome and genome projects independently. In the subsequent collaboration, both groups worked closely on every aspect of the project. Chicago group: C.B.A., Z.Y.W., J.R.P. and C.W.R.; OIST/Berkeley group: O.S., T.M., E.E.-G., S.B. and D.S.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Genome and transcriptome sequence reads have been deposited in the SRA as BioProjects PRJNA270931 and PRJNA285380. A browser of this genome assembly is available at (http://octopus.metazome.net/).
Extended data figures and tables
Extended Data Figure 1 RNA editing in octopus.
a, Approximate maximum likelihood tree of adenosine deaminases acting on RNA (ADARs) in bilaterians. ADAR1, ADAR2, ADAR-like/ADAD, and ADAT (tRNA-specific adenosine deaminase) were identified in Hsa, Mmu, Cin, Dme, Cte, Lgi, D. opalescens (Dop54), and Obi with Shimodaira–Hasegawa-like support indicated at the nodes. b, O. bimaculoides ADAR1, ADAR2 and ADAR-like proteins contain one or two double-stranded RNA binding domains (dsRBD) as well as an adenosine deaminase domain. ADAR1 also has a z-alpha domain. c, Expression profiles of the three ADAR genes found in 12 O. bimaculoides tissues by RNA-seq profiling. d, DNA–RNA differences in O. bimaculoides show prominent A-to-G changes. Histogram illustrates the number of DNA–RNA differences detected between coding sequences in the genome and 12 O. bimaculoides transcriptomes after filtering out polymorphisms identified in genomic sequencing. Differences were binned by the type of change (see key) in the direction of transcription. A-to-G changes are the most prevalent, particularly in neural tissues and during development, paralleling the expression of octopus ADARs in c. Other types of changes were also detected at lower levels, possibly resulting from uncharacterized polymorphisms.
Extended Data Figure 2 Local arrangement of Hox gene complement in O. bimaculoides and selected bilaterians.
At the top, the four compact Hox clusters of H. sapiens and the single B. floridae cluster are depicted. The D. melanogaster Hox complex is split into two clusters. We included genes in the D. melanogaster locus that are homologues of Hox genes but have lost their homeotic function, such as fushi tarazu (ftz), bicoid, zen and zen2 (the latter three are represented as overlapping boxes). Hox genes in C. teleta are found on three scaffolds17. L. gigantea has a single cluster with the full known lophotrochozoan gene complement. In O. bimaculoides many of the scaffolds are several hundred kb long, and no two Hox genes are on the same scaffold. The positions of O. bimaculoides genes approximate their locations on scaffolds. Dashed lines indicate that the scaffold continues beyond what is shown. Scaffold length is depicted to scale with size noted on the left. Genes are positioned to illustrate orthology, which is also highlighted by colour.
Extended Data Figure 3 Gene complement and gene architecture evolution in metazoans.
a, Principal component analysis of gene family counts. O. bimaculoides highlighted in green. Deuterostomes are indicated in blue, ecdysozoans in red, lophotrochozoans in green, and sponges and cnidarians in orange. Xtr, Xenopus tropicalis; Gga, Gallus gallus; Tca, Tribolium castaneum; Dpu, Daphnia pulex; Isc, Ixodes scapularis; Ava, Adineta vaga; Spu, S. purpuratus; Hma, Hydra magnipapillata; Adi, Acropora digitifera. For methods, see Supplementary Note 7.4. b–d, MrBayes55 tree (constrained topology) on binary characters of presence or absence of Pfam domain architectures (b), introns (c), or indels (d); scale bar represents estimated changes per site. For methods, see Supplementary Note 7.3.
Extended Data Figure 4 Protocadherin genes within a genomic cluster are similar in sequence and sites of expression.
a, Expression profile of the 31 protocadherin genes located on Scaffold 30672 in 12 octopus transcriptomes. Over three-quarters of the protocadherins are highly expressed throughout central brain, OL and ANC, while the others show more mixed distributions. b, Phylogenetic tree highlighting Scaffold 30672 protocadherins in grey bars. c, Expression profile of the 17 protocadherin genes located on Scaffold 9600. Almost all of these protocadherins are most highly expressed in nervous tissues, with the exception of Ocbimv220039316m, which is most highly expressed in the St15 sample. d, Phylogenetic tree highlighting Scaffold 9600 protocadherins in grey bars. As seen in b, protocadherins of the same scaffold tend to cluster together on the tree. Order of the genes in the heat maps (a, c) follows the ordering on the corresponding scaffold.
Extended Data Figure 5 Expansion of interleukin 17 (IL17)-like genes.
a, Phylogenetic tree of interleukin genes in Obi, Cte, Cgi, and Lgi. Mammalian IL1A, IL1B, and IL7 used as outgroups. Human and mouse IL17s branch from other members of the IL family. Octopus ILs (as well as all identified invertebrate ILs) group with the mammalian IL17 branch and are named ‘IL17-like’. The 31 octopus genes are distributed across 5 scaffolds: scaffold A (Obi_A), 23 members; scaffold B (Obi_B), 4 members; scaffold C (Obi_C), 2 members; scaffolds D (Obi_D) and E (Obi_E), 1 member each. b, Expression profile of 31 octopus IL17-like genes. Heat map rows are arranged by order on each scaffold. Blank rows indicate genes not expressed in our transcriptomes. The 27 genes found in our transcriptomes have strong expression in the suckers and skin. The scaffold C genes are enriched in the PSG and the Scaffold D gene is enriched in the viscera. c, Conserved cysteine residues in human IL17 and invertebrate IL17-like proteins. The human IL17 proteins share a conserved cysteine motif comprising 4 cysteine residues, which may form interchain disulfide bonds and facilitate dimerization56. Octopus IL17-like proteins also contain this four-cysteine motif, highlighted in yellow. One octopus sequence encodes only 3 of these highly conserved cysteine residues. These four cysteines are also present to varying degrees in Lottia, Capitella and Crassostrea sequences. Two additional conserved cysteine residues were found in the octopus sequences and are highlighted in red. The first cysteine residue is found in all invertebrate sequences examined, and none of the mammalian IL17 sequences.
Extended Data Figure 6 G-protein-coupled receptors.
GPCRs, also known as 7-transmembrane (7TM) or serpentine receptors, form a large superfamily that activates intracellular second messenger systems upon ligand binding. This figure considers a subset of the 329 GPCRs we identified in O. bimaculoides. The full complement of GPCRs is presented in Supplementary Note 8.5. a, b, As reported for other lophotrochozoan genomes, the octopus genome contains chemosensory-like GPCRs; 74 GPCRs are similar to the Aplysia chemosensory GPCRs57 and 11 GPCRs are similar to vertebrate olfactory receptors. c, We identified 4 opsins in the octopus genome (from top to bottom): rhodopsin, rhabdomeric opsin, peropsin, and retinochrome. d, The octopus class F GPCRs comprises 6 genes: 5 Frizzled genes and 1 Smoothened gene (*). e, Thirty octopus genes show similarity to vertebrate adhesion GPCRs.
Extended Data Figure 7 O. bimaculoides acetylcholine receptor (AChR) subunits.
a, Phylogenetic tree of AChR subunit genes identified in Hsa, Mmu, Dme, Cte, Lgi and Obi. Black asterisk indicates a Dme sequence that groups with alpha 1-4-like subunits despite lacking two defining cysteine residues. b, Expression profiles of octopus AChR subunits. Genes ordered as in the tree (a), starting from the grey arrow and continuing anticlockwise. Putative non-ACh-binding subunits are highly expressed in the suckers. One sequence was not detected in our transcriptome data sets. In a and b, red asterisks indicate subunits with the substitution known to confer anionic permissivity58. c, Divergent octopus subunits lack nearly all residues necessary for ACh binding. Alignment of sequence flanking the cysteine loop (yellow) of the L. stagnalis ACh-binding protein (Lst_AchBP), the human and octopus alpha-7 receptor subunits (Hsa_AchR7, Obi_10697+), and the 23 divergent AChR subunits. Essential ACh-binding residues on the primary (pink) and complementary (blue) side of the ligand-binding domain are indicated26, with conservative substitutions in a lighter shade. Outside of the binding residues, residues shared between the alpha-7 subunits are shaded in light grey, with bold letters for conservative substitutions.
Extended Data Figure 8 Active transposable elements and gene expression specificity.
a, Transposable element expression across 12 tissues. b, Correlation between the total transposable element (TE) load (in bp) in the 5 kb regions flanking the gene and the fraction of genes with tissue-specific expression (defined as having at least 75% of expression in a single tissue; see Source Data file for this figure). P value indicates the F-statistic for the significance of linear regression (H0: r2 = 0), with tissues with a P value ≤0.05 indicated in pink.
Extended Data Figure 9 Synteny dynamics in octopus and the effect of transposable element (TE) expansions.
a, Circos plot showing shared synteny across 6 genomes. Individual scaffolds are plotted according to bp length; scaffolds with no synteny are merged together (lighter arcs). Despite the large size of the octopus genome, only a small proportion of the scaffolds show synteny. b, Synteny reduction in octopus quantified based on synteny inference using gene families with at least one representative in human, amphioxus, Capitella, Helobdella, Octopus, Lottia, Crassostrea, Drosophila, and Nematostella. Drosophila, Helobdella and Octopus show the highest synteny loss rates. Branch lengths, estimated with MrBayes55, reflect extent of local genome rearrangement (Supplementary Note 6). c, Enrichment of overall and specific TE classes (base pairs masked) around genes from ancient bilaterian synteny blocks, including those absent in octopus (see key). Asterisks indicate Mann–Whitney U-test with P value <0.02. d, Transposable element insertion history (Jukes–Cantor distance adjusted, see text) into the vicinity of genes from ‘lost’ synteny blocks. Note that only one SINE peak is present; a more recent peak (visible in ‘All genomic SINEs’) cannot be recovered from those insertions.
Extended Data Figure 10 Cephalopod phylogeny and novelties.
a, Whole-genome-derived phylogeny of molluscs and select other phyla showing the relative position of octopus at the base of the coleoid cephalopods. For methods see Supplementary Note 7.1. Members of the cephalopod class are indicated in blue, scale indicates number of substitutions per site. b, Phylogenetic tree of reflectin genes. Reflectins are cephalopod-specific genes that allow for rapid and reversible changes in iridescence. Six reflectin genes were identified in the octopus genome. c, d, Novel gene expression across multiple tissues. Bars depict all cephalopod novelties; dark grey indicates sequences with no similarity to non-cephalopod genes using HMM searches (see Source Data for this figure). c, Counts of tissue-specific novelties in a given tissue. d, Proportion of expression of novel genes versus total expression in individual tissues. CNS (central nervous system) combines Supra, Sub, OL and ANC expression data.
Extended Data Figure 11 Phylogenetic tree of cadherin genes.
This is a larger image of Fig. 2a. Phylogenetic tree of cadherin genes in Hsa (red), Dme (orange), Nematostella vectensis (mustard yellow), Amphimedon queenslandica (yellow), Cte (green), Lgi (teal), Obi (blue), and Saccoglossus kowalevskii (purple). I, Type I classical cadherins; II, calsyntenins; III, octopus protocadherin expansion (168 genes); IV, human protocadherin expansion (58 genes); V, dachsous; VI, fat-like; VII, fat; VIII, CELSR; IX, Type II classical cadherins. Asterisk denotes a novel cadherin with over 80 extracellular cadherin domains found in Obi and Cte.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data, Supplementary Tables, Supplementary Figures and additional references (See Contents for details). (PDF 11559 kb)
Supplementary Figure - Metazoan C2H2 superfamily tree
Approximate maximum likelihood tree of metazoan C2H2-ZNFs. The tree was built using a near-complete subset of transcriptome-confirmed C2H2-ZNF sequences (1,452/1,790) from octopus (dark blue), 539 from human (red), 388 from mouse (pink), 159 from zebrafish (purple), 7 from A. queenslandica (yellow), 88 from A. californica (aqua), 24 from C. intestinalis (dark purple), 65 from S. kowalevskii (violet), 44 from T. castaneum (orange), 39 from N. vectensis (mustard), 87 from L. gigantea (teal), and 72 from C. teleta (green). (PDF 7114 kb)
Supplementary Figure - Metazoan GPCR superfamily tree
Approximate maximum likelihood tree of bilaterian GPCRs. The tree was built using the 329 GPCR sequences from octopus (dark blue), 185 from human (red), 160 from mouse (pink), 226 from zebrafish (magenta), 204 from S. kowalevskii (purple), 125 from L.gigantea (aqua), 258 from C. teleta (green), and 87 from A. californica (grey-blue). For clarity, only a subset of vertebrate olfactory receptors were used. The octopus Class A rhodopsins are centered around 7 o'clock. (PDF 229 kb)
Octopus bimaculoides: Adaptive coloration
The ability to camouflage is present throughout the animal kingdom, but cephalopods uniquely achieve adaptive coloration. Three different structures in the skin contribute to the color of an octopus: chromatophores, iridophores, and leucophores. The reflectin family of proteins reversibly regulates iridescence. Muscles in the skin allow octopuses to alter texture and disrupt the visual boundaries of the body. Most importantly, this complex behavior happens very quickly, allowing octopuses to change their appearance almost instantaneously. The O. bimaculoides genome and transcriptome is reported by Albertin and Simakov et al. Credit: Z. Yan Wang, C. Ragsdale, J. Reynolds, and Schadenfreude the octopus. (MP4 36989 kb)
Octopus bimaculoides: Arms and Suckers
Octopuses are perhaps most easily recognized by their eight appendages. The arms are strong muscular hydrostats, each covered with hundreds of suckers that allow octopuses to exert powerful suction forces. The flexibility and strength of octopus arms is a unique morphological innovation in the animal kingdom. The O. bimaculoides genome and transcriptome is reported by Albertin and Simakov et al. Credit: Z. Yan Wang, C. Ragsdale, J. Reynolds, and Schadenfreude the octopus. (MP4 36819 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Albertin, C., Simakov, O., Mitros, T. et al. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224 (2015). https://doi.org/10.1038/nature14668
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14668
This article is cited by
-
Gene modelling and annotation for the Hawaiian bobtail squid, Euprymna scolopes
Scientific Data (2024)
-
Oldest known animal sex chromosome evolved in octopuses 380 million years ago
Nature (2024)
-
Chromosome-level genome assembly of the deep-sea snail Phymorhynchus buccinoides provides insights into the adaptation to the cold seep habitat
BMC Genomics (2023)
-
Gene losses, parallel evolution and heightened expression confer adaptations to dedicated cleaning behaviour
BMC Biology (2023)
-
Hologenome analysis reveals independent evolution to chemosymbiosis by deep-sea bivalves
BMC Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
