
Abstract
G protein-coupled receptors (GPCRs) allosterically activate heterotrimeric G proteins and trigger GDP release. Given that there are ∼800 human GPCRs and 16 different Gα genes, this raises the question of whether a universal allosteric mechanism governs Gα activation. Here we show that different GPCRs interact with and activate Gα proteins through a highly conserved mechanism. Comparison of Gα with the small G protein Ras reveals how the evolution of short segments that undergo disorder-to-order transitions can decouple regions important for allosteric activation from receptor binding specificity. This might explain how the GPCR–Gα system diversified rapidly, while conserving the allosteric activation mechanism.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
Vetter, I. R. & Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 (2001)
Leipe, D. D., Wolf, Y. I., Koonin, E. V. & Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317, 41–72 (2002)
Rojas, A. M., Fuentes, G., Rausell, A. & Valencia, A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J. Cell Biol. 196, 189–201 (2012)
Anantharaman, V., Abhiman, S., de Souza, R. F. & Aravind, L. Comparative genomics uncovers novel structural and functional features of the heterotrimeric GTPase signaling system. Gene 475, 63–78 (2011)
Rasmussen, S. G. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555 (2011)
Chung, K. Y. et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477, 611–615 (2011)
Preininger, A. M., Meiler, J. & Hamm, H. E. Conformational flexibility and structural dynamics in GPCR-mediated G protein activation: a perspective. J. Mol. Biol. 425, 2288–2298 (2013)
Oldham, W. M., Van Eps, N., Preininger, A. M., Hubbell, W. L. & Hamm, H. E. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nature Struct. Mol. Biol. 13, 772–777 (2006)
Westfield, G. H. et al. Structural flexibility of the Gαs α-helical domain in the β2-adrenoceptor Gs complex. Proc. Natl Acad. Sci. USA 108, 16086–16091 (2011)
Alexander, N. S. et al. Energetic analysis of the rhodopsin-G-protein complex links the α5 helix to GDP release. Nature Struct. Mol. Biol. 21, 56–63 (2014)
Neves, S. R., Ram, P. T. & Iyengar, R. G protein pathways. Science 296, 1636–1639 (2002)
Chothia, C. & Lesk, A. M. Helix movements and the reconstruction of the haem pocket during the evolution of the cytochrome c family. J. Mol. Biol. 182, 151–158 (1985)
Süel, G. M., Lockless, S. W., Wall, M. A. & Ranganathan, R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature Struct. Biol. 10, 59–69 (2003)
del Sol, A., Fujihashi, H., Amoros, D. & Nussinov, R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2, (2006)
Kornev, A. P., Haste, N. M., Taylor, S. S. & Eyck, L. F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl Acad. Sci. USA 103, 17783–17788 (2006)
Zhang, X., Perica, T. & Teichmann, S. A. Evolution of protein structures and interactions from the perspective of residue contact networks. Curr. Opin. Struct. Biol. 23, 954–963 (2013)
Deupi, X. et al. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl Acad. Sci. USA 109, 119–124 (2012)
Standfuss, J. et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471, 656–660 (2011)
Choe, H. W. et al. Crystal structure of metarhodopsin II. Nature 471, 651–655 (2011)
Scheerer, P. et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 (2008)
Slessareva, J. E. et al. Closely related G-protein-coupled receptors use multiple and distinct domains on G-protein α-subunits for selective coupling. J. Biol. Chem. 278, 50530–50536 (2003)
Oldham, W. M. & Hamm, H. E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nature Rev. Mol. Cell Biol. 9, 60–71 (2008)
Dratz, E. A. et al. NMR structure of a receptor-bound G-protein peptide. Nature 363, 276–281 (1993)
Tompa, P., Davey, N. E., Gibson, T. J. & Babu, M. M. A million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 (2014)
Sun, D. et al. Probing Gαi1 protein activation at single amino acid resolution. Nature Struct. Mol. Biol (in the press)
Mnpotra, J. S. et al. Structural basis of G protein-coupled receptor-Gi protein interaction: formation of the cannabinoid CB2 receptor-Gi protein complex. J. Biol. Chem. 289, 20259–20272 (2014)
Hanson, P. I. & Whiteheart, S. W. AAA+ proteins: have engine, will work. Nature Rev. Mol. Cell Biol. 6, 519–529 (2005)
Nesbit, M. A. et al. Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. N. Engl. J. Med. 368, 2476–2486 (2013)
Iiri, T., Herzmark, P., Nakamoto, J. M., van Dop, C. & Bourne, H. R. Rapid GDP release from Gsα in patients with gain and loss of endocrine function. Nature 371, 164–168 (1994)
Li, D. et al. Autosomal dominant hypoparathyroidism caused by germline mutation in GNA11: phenotypic and molecular characterization. J. Clin. Endocrinol. Metab. 99, E1774–E1783 (2014)
de la Vega, M., Burrows, J. F. & Johnston, J. A. Ubiquitination: Added complexity in Ras and Rho family GTPase function. Small GTPases 2, 192–201 (2011)
Oldham, W. M. & Hamm, H. E. Structural basis of function in heterotrimeric G proteins. Q. Rev. Biophys. 39, 117–166 (2006)
Cui, Q. & Karplus, M. Allostery and cooperativity revisited. Protein Sci. 17, 1295–1307 (2008)
Brown, C. J., Johnson, A. K., Dunker, A. K. & Daughdrill, G. W. Evolution and disorder. Curr. Opin. Struct. Biol. 21, 441–446 (2011)
Tanaka, T. et al. α helix content of G protein or subunit is decreased upon activation by receptor mimetics. J. Biol. Chem. 273, 3247–3252 (1998)
Natochin, M., Moussaif, M. & Artemyev, N. O. Probing the mechanism of rhodopsin-catalyzed transducin activation. J. Neurochem. 77, 202–210 (2001)
Marin, E. P., Krishna, A. G. & Sakmar, T. P. Disruption of the α5 helix of transducin impairs rhodopsin-catalyzed nucleotide exchange. Biochemistry 41, 6988–6994 (2002)
Marin, E. P., Krishna, A. G. & Sakmar, T. P. Rapid activation of transducin by mutations distant from the nucleotide-binding site: evidence for a mechanistic model of receptor-catalyzed nucleotide exchange by G proteins. J. Biol. Chem. 276, 27400–27405 (2001)
Flicek, P. et al. Ensembl 2014. Nucleic Acids Res. 42, D749–D755 (2014)
Kasprzyk, A. BioMart: driving a paradigm change in biological data management. Database 2011, bar049 (2011)
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990)
Johnston, C. A. et al. Structural determinants underlying the temperature-sensitive nature of a Gα mutant in asymmetric cell division of Caenorhabditis elegans . J. Biol. Chem. 283, 21550–21558 (2008)
Jia, M. et al. Crystal structures of the scaffolding protein LGN reveal the general mechanism by which GoLoco binding motifs inhibit the release of GDP from Gαi. J. Biol. Chem. 287, 36766–36776 (2012)
Magrane, M. & Consortium, U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011, bar009 (2011)
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
Ruan, J. et al. TreeFam: 2008 Update. Nucleic Acids Res. 36, D735–D740 (2008)
Dereeper, A. et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 (2008)
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010)
Chevenet, F., Brun, C., Banuls, A. L., Jacq, B. & Christen, R. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinform. 7, 439 (2006)
Altenhoff, A. M., Schneider, A., Gonnet, G. H. & Dessimoz, C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 39, D289–D294 (2011)
Vilella, A. J. et al. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335 (2009)
Pei, J., Sadreyev, R. & Grishin, N. V. PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19, 427–428 (2003)
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006)
Ballesteros, J. A. & Weinstein, H. Receptor Molecular Biology Vol. 25 (Elsevier, 1995)
Velankar, S. et al. SIFTS: Structure Integration with Function, Taxonomy and Sequences resource. Nucleic Acids Res. 41, D483–D489 (2013)
Heinig, M. & Frishman, D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32, W500–W502 (2004)
Konagurthu, A. S., Whisstock, J. C., Stuckey, P. J. & Lesk, A. M. MUSTANG: a multiple structural alignment algorithm. Proteins 64, 559–574 (2006)
Noel, J. P., Hamm, H. E. & Sigler, P. B. The 2.2 Å crystal structure of transducin-α complexed with GTPγS. Nature 366, 654–663 (1993)
Doncheva, N. T., Klein, K., Domingues, F. S. & Albrecht, M. Analyzing and visualizing residue networks of protein structures. Trends Biochem. Sci. 36, 179–182 (2011)
Grant, B. J., Rodrigues, A. P., ElSawy, K. M., McCammon, J. A. & Caves, L. S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696 (2006)
Babu, M. M. NCI: A server to identify non-canonical interactions in protein structures. Nucleic Acids Res. 31, 3345–3348 (2003)
Morikawa, T. et al. Crystallization and preliminary X-ray crystallographic analysis of the receptor-uncoupled mutant of Gα1. Acta Crystallogr. F. 63, 139–141 (2007)
Lopes, C. T. et al. Cytoscape Web: an interactive web-based network browser. Bioinformatics 26, 2347–2348 (2010)
Shannon, P. T., Grimes, M., Kutlu, B., Bot, J. J. & Galas, D. J. RCytoscape: tools for exploratory network analysis. BMC Bioinform. 14, 217 (2013)
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007)
Dosztanyi, Z., Csizmok, V., Tompa, P. & Simon, I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 (2005)
Vohra, S. & Biggin, P. C. Mutationmapper: a tool to aid the mapping of protein mutation data. PLoS ONE 8, e71711 (2013)
Forbes, S. A. et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 (2015)
Stenson, P. D. et al. The Human Gene Mutation Database: 2008 update. Genome Med 1, 13 (2009)
Sun, D. et al. AAscan, PCRdesign and MutantChecker: a suite of programs for primer design and sequence analysis for high-throughput scanning mutagenesis. PLoS ONE 8, e78878 (2013)
Maeda, S. et al. Crystallization scale preparation of a stable GPCR signaling complex between constitutively active rhodopsin and G-protein. PLoS ONE 9, e98714 (2014)
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000)
Pai, E. F. et al. Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 Å resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 9, 2351–2359 (1990)
Milburn, M. V. et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247, 939–945 (1990)
Boriack-Sjodin, P. A., Margarit, S. M., Bar-Sagi, D. & Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 394, 337–343 (1998)
Acknowledgements
We thank U. F. Lang, C. Chothia, R. Weatheritt, S. Balaji, H. Harbrecht, A. Morgunov, G. Murshudov and N. S. Latysheva for their comments on this work. This work was supported by the Medical Research Council (MC_U105185859; M.M.B., T.F., M.K., A.J.V. and C.N.J.R.; MC_U105197215; C.G.T.), the Swiss National Science Foundation grants 141898, 133810, 31-135754 (D.B.V.), the MRC Centenary Award (A.J.V.), the AFR scholarship from the Luxembourg National Research Fund (C.N.J.R.) and the Boehringer Ingelheim Fond (T.F.). M.M.B. is also a Lister Institute Research Prize Fellow.
Author information
Authors and Affiliations
Contributions
T.F. collected data, wrote scripts and performed all the analysis. C.N.J.R. helped with data analysis and writing the scripts for structure analysis with T.F. A.J.V. helped T.F. with data analysis. D.S. and D.B.V. contributed experimental results on alanine scanning. M.K. prepared the CGN webserver. C.G.T. helped with aspects of data interpretation. T.F. and M.M.B. designed the project, analysed the results and wrote the manuscript. All authors read and provided their comments on the draft. M.M.B. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Human paralogue reference alignment for common Gα numbering system.
a, Reference alignment of all canonical human Gα paralogues. The domain (D), consensus secondary structure (S) and position in the SSE of the human reference alignment (P) are shown on top of the alignment. b, Reference table of the definitions of SSEs used in the CGN nomenclature.
Extended Data Figure 2 Energy estimation of the GPCR–Gα residue contributions and Gα disorder propensity.
a, Energy contribution of single interface residues to the Gαs–β2AR complex calculated with FoldX (T = 298K, pH = 7.0, ionic strength = 0.05M). Conserved Gα residues (blue sequence logo) that were identified to form receptor–Gα inter-protein contacts with conserved GPCR residues (red sequence logo) are shown. The contact network between residues of the β2AR and Gαs is shown (red, conserved receptor residue; blue, conserved Gα residue; grey, variable residues; spheres represent Cα positions and links represent non-covalent contact). b, Consensus disorder plot for all Gα proteins. The mean value of the disorder propensity of all full-length Gα sequences (561 sequences; homologous to all 16 human Gα proteins) is shown as a black line; the standard deviation at each position is shown as light red ribbon. The colour tone of the line indicates the number of gaps at an aligned position (black, no gaps). The left inset shows the disorder propensity of H1. The right inset highlights that H5 is highly structured in its N terminus, and has increased disorder propensity towards the C terminus, which is in agreement with the missing electron density in the 79 structures.
Extended Data Figure 3 Rewiring of consensus contacts between conserved Gα residues upon receptor binding.
CGN numbers and sequence logo for consensus contacts within Gα in the inactive state (left) and GPCR-bound state (right) are shown. Receptor residues are shown in red; H5 residues in dark blue; H1 residues in light blue; and GDP in green. The domains are highlighted with a blue background (G-domain darker blue, H-domain light blue). This figure highlights the most important consensus residue contacts between conserved residues. Additional contacts in the right lobe are discussed in the Supplementary Note and in ref. 25. For a full list of residue contacts, please refer to Supplementary Data.
Extended Data Figure 4 Details of helix H1 linking H5, GDP and the H-domain.
a, This figure expands Fig. 4 from the main text to provide residue-level details of the role of helix H1. Residues forming contacts with H5 are shown in blue, with the H-domain in light blue and with GDP in green. Non-covalent consensus contacts between universally conserved residues at the SSE level (left) and per residue-level (centre). Lines denote non-covalent contacts between residues. The degree of conservation is shown as sequence logo. Residues are numbered according to the CGN. Helix H1 is almost 100% conserved across all 16 Gα types and forms three structural motifs for interactions with H5, the H-domain and GDP (right). b, Average per residue energy contribution to Gα protein stability as calculated from 79 structures from all four Gα subfamilies in the non-receptor-bound signalling states using FoldX (T = 298K, pH = 7.0, ionic strength = 0.05M). The average energy contribution is shown as dots, the standard deviation as bars. c, Per residue detail of Gα-GDP and Gα-GSP (non-hydrolysable GTP analogue) consensus contacts. The bar-plot shows the frequency of finding a contact mediated by topologically equivalent positions with GDP/GSP. Number of side-chain and main-chain contacts are shown as dark grey and light grey bars, respectively. The degree of conservation of contacting residues (calculated from the 561 complete Gα sequences) is represented in the right panel and the consensus sequence for each position is shown.
Extended Data Figure 5 Conserved structural motifs of Gα and known disease and engineered mutations.
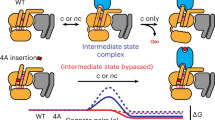
a, A universally conserved cluster of π–π and hydrophobic interactions between S2 (PheG.S2.6) and S3 (PheG.S3.3), H1 (MetG.H1.8 and HisG.H1.12) and H5 (PheG.H5.8) links H5 and H1 in the absence of the receptor. Upon receptor binding, residues within this motif (PheG.H5.8 and PheG.S3.3) interact with the conserved Pro and Leu of ICL2 of the receptor as has been shown for Gαs (3sn6) and Gαi (ref. 26). Interrupting the contacts between H5 and H1 seems to be the trigger for transmitting the signal of GPCR binding to helix H1 (which interacts with GDP and the H-domain.) The only conserved residue contact between the H-domain and the G-domain that is not in the hinge region is formed by a universally conserved salt bridge (H-domain ionic latch) between the very N-terminal end of HG of the G-domain (LysG.s5hg.1) and the loop connecting HD and HE in the H domain (AspH.hdhe.5). The hinge region is formed by H1, the loop between H1 and HA, and HF. H1 interacts via (1) a cation–π interaction mediated by a universally conserved residue with the loop connecting H1 and HA (LysG.H1.6 and TyrG.h1ha.4) and (2) a hydrophobic interaction with HF (LysG.H1.9 and LeuH.HF.5). b, Disease and engineered mutations that can be explained by the universal Gα activation mechanism mapped on a Gα protein. Cα position of residues are shown as spheres; mutations at green positions cause spontaneous GDP release by interrupting consensus contacts between conserved residues, thereby ‘mimicking’ the effect of receptor binding to Gα. Pink positions have also been reported to cause disease by constitutively activating Gα. Insertion of an Ala4 or Gly5 after the yellow position separate the H5 transmission and interface module, thereby allowing GPCR binding without triggering GDP release.
Extended Data Figure 6 Helix H5–H1 interaction in Gα provides the allosteric GEF activation mechanism.
a, Schematic representation of structural motifs on H1 that are shared or unique to Gα and Ras. While the part of H1 with the phosphate-binding motif is conserved across both protein families, the C-terminal part is conserved only in Gα. H1 in Gα has three additional residues that allow for extensive residue contacts between H1 and H5. In Ras, these interactions are missing and H5 and H1 are both 3 residues shorter. The consensus sequence and secondary structure of equivalent residues of H1 in Gα and Ras is also depicted. b, Comparison of the residue contact network between topologically equivalent residues in H5 and H1 in the corresponding inactive GDP-bound states of Gα (1got) and Ras (4q21). The weight of the link between SSEs denotes the number of atomic contacts. c, Sequence alignments of H1 and H5 of human Gα and Ras paralogues. The sequence alignment was obtained based on cross-referencing the alignments using the structures of Gα and Ras.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-2, Supplementary Notes and additional references. (PDF 2276 kb)
Supplementary Table 1
This file shows the Gα structure and sequence data. (XLSX 69 kb)
Supplementary Table 2
This file shows the Common Gα reference system data. (XLSX 109 kb)
Supplementary Table 3
This file shows engineered and natural mutations in G proteins reported in literature. (XLSX 52 kb)
Supplementary Data
This zipped file contains 5 files which comprise: Ortholog alignments; Visualization of consensus networks (≧75% consensus score) of the four signaling states with residue conservation represented as B-factor; Visualization of the GPCR-G protein interface in the 3sn6 crystal structure; Gα -Ras alignment; and Consensus residue interaction networks with sequence conservation for Gα and GPCRGα interface residue interaction networks for Gs-β 2-adrenergic receptor and Gt peptides-rhodopsin. (ZIP 3645 kb)
Rights and permissions
About this article

Cite this article
Flock, T., Ravarani, C., Sun, D. et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179 (2015). https://doi.org/10.1038/nature14663
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14663
This article is cited by
-
Dissecting the genetic landscape of GPCR signaling through phenotypic profiling in C. elegans
Nature Communications (2023)
-
Two-step structural changes in M3 muscarinic receptor activation rely on the coupled Gq protein cycle
Nature Communications (2023)
-
Ligand recognition mechanism of the human relaxin family peptide receptor 4 (RXFP4)
Nature Communications (2023)
-
GPCRome-wide analysis of G-protein-coupling diversity using a computational biology approach
Nature Communications (2023)
-
The role of G protein conformation in receptor–G protein selectivity
Nature Chemical Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
