
Abstract
TP53, a well-known tumour suppressor gene that encodes p53, is frequently inactivated by mutation or deletion in most human tumours1,2. A tremendous effort has been made to restore p53 activity in cancer therapies3,4,5,6,7. However, no effective p53-based therapy has been successfully translated into clinical cancer treatment owing to the complexity of p53 signalling. Here we demonstrate that genomic deletion of TP53 frequently encompasses essential neighbouring genes, rendering cancer cells with hemizygous TP53 deletion vulnerable to further suppression of such genes. POLR2A is identified as such a gene that is almost always co-deleted with TP53 in human cancers. It encodes the largest and catalytic subunit of the RNA polymerase II complex, which is specifically inhibited by α-amanitin8,9. Our analysis of The Cancer Genome Atlas (TCGA) and Cancer Cell Line Encyclopedia (CCLE) databases reveals that POLR2A expression levels are tightly correlated with its gene copy numbers in human colorectal cancer. Suppression of POLR2A with α-amanitin or small interfering RNAs selectively inhibits the proliferation, survival and tumorigenic potential of colorectal cancer cells with hemizygous TP53 loss in a p53-independent manner. Previous clinical applications of α-amanitin have been limited owing to its liver toxicity10. However, we found that α-amanitin-based antibody–drug conjugates are highly effective therapeutic agents with reduced toxicity11. Here we show that low doses of α-amanitin-conjugated anti-epithelial cell adhesion molecule (EpCAM) antibody lead to complete tumour regression in mouse models of human colorectal cancer with hemizygous deletion of POLR2A. We anticipate that inhibiting POLR2A will be a new therapeutic approach for human cancers containing such common genomic alterations.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Change history
25 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41586-021-03664-3
References
Petitjean, A. et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629 (2007)
Vazquez, A. et al. The genetics of the p53 pathway, apoptosis and cancer therapy. Nature Rev. Drug Discov. 7, 979–987 (2008)
Cheok, C. F. et al. Translating p53 into the clinic. Nature Rev. Clin. Oncol. 8, 25–37 (2011)
Lane, D. P., Cheok, C. F. & Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2, a001222 (2010)
Chène, P. Inhibiting the p53–MDM2 interaction: an important target for cancer therapy. Nature Rev. Cancer 3, 102–109 (2003)
Wade, M., Li, Y. C. & Wahl, G. M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nature Rev. Cancer 13, 83–96 (2013)
Haupt, S. & Haupt, Y. Manipulation of the tumor suppressor p53 for potentiating cancer therapy. Semin. Cancer Biol. 14, 244–252 (2004)
Bensaude, O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2, 103–108 (2011)
Lindell, T. J. et al. Specific inhibition of nuclear RNA polymerase II by α-amanitin. Science 170, 447–449 (1970)
Letschert, K. et al. Molecular characterization and inhibition of amanitin uptake into human hepatocytes. Toxicol. Sci. 91, 140–149 (2006)
Moldenhauer, G. et al. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J. Natl. Cancer Inst. 104, 622–634 (2012)
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability–an evolving hallmark of cancer. Nature Rev. Mol. Cell Biol. 11, 220–228 (2010)
Nijhawan, D. et al. Cancer vulnerabilities unveiled by genomic loss. Cell 150, 842–854 (2012)
Muller, F. L. et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488, 337–342 (2012)
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014)
Toledo, F. & Wahl, G. M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nature Rev. Cancer 6, 909–923 (2006)
Wang, T. et al. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014)
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013)
Derheimer, F. A. et al. RPA and ATR link transcriptional stress to p53. Proc. Natl Acad. Sci. USA 104, 12778–12783 (2007)
Lu, J. et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 23, 171–185 (2013)
Faulstich, H. & Fiume, L. Protein conjugates of fungal toxins. Methods Enzymol. 112, 225–237 (1985)
Went, P. T. et al. Frequent EpCam protein expression in human carcinomas. Hum. Pathol. 35, 122–128 (2004)
Pecot, C. V. et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol. Cancer Ther. 13, 2876–2885 (2014)
Goldstein, I. et al. Understanding wild-type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 18, 2–11 (2011)
Liu, Y. et al. Kaposi’s sarcoma-associated herpesvirus-encoded microRNA miR-K12-11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J. Virol. 86, 1372–1381 (2012)
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013)
Guschin, D. Y. et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 (2010)
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols 8, 2281–2308 (2013)
Acknowledgements
We thank F. Zhang and I. J. Fidler for technical support in orthotopic tumour experiments. We thank L. Huang and M. Bar-Eli for lentivirus production. This work was supported by grants to X.L. (National Institutes of Health (NIH) R01 CA136549, MD Anderson Moon Shots Program) and to A.K.S. (NIH U54 CA151668).
Author information
Authors and Affiliations
Contributions
Y.L., X.Z. and X.L. directed the work and wrote the manuscript. C.H. and G.W. generated vectors and performed cell-based assays. X.H. provided technical guidance on CRISPR/Cas9 and gene knockout experiments. D.M.M. provided clinical tissue samples and technical guidance. D.J., C.R.-A. and G.L.-B. produced siRNA–DOPC. P.H.R. carried out FISH experiments. J.A. and A.P. provided α-amanitin-conjugated antibodies and technical guidance in xenograft tumour experiments. L.M.E., X.H., C.I. and A.K.S. helped to analyse data. All authors discussed the results and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.A. and A.P. are employees of Heidelberg Pharma GmbH.
Extended data figures and tables
Extended Data Figure 1 Expression of POLR2A correlates with its gene copy number in human colon tumours.
a, Top, double-colour FISH analysis using a probe for chromosome 17 centromere (green) and a locus-specific probe for POLR2A (red) on human colon tissue microarray. Bottom, immunohistochemical staining of POLR2A in the corresponding tissue samples. Hemizygous loss of the POLR2A gene was determined, and the results are shown in Supplementary Table 2. b, Quantification of POLR2A expression in human colon normal (n = 7), POLR2Aneutral (n = 43) or POLR2Aloss (n = 29) tumour tissue samples. Error bars, s.d. c, Protein levels of POLR2A and β-actin in matched normal and CRC tissue samples.
Extended Data Figure 2 Expression of TP53 is not associated with its gene copy number.
a, b, Scatterplots of TP53 copy number versus protein expression (a) or mRNA expression (b) in colorectal tumours in TCGA database. Pearson correlation coefficients (r) and P values are displayed. c, Relative mRNA expression of TP53 in human CRC cell lines (normalized to that in the HCT116 cell line). Data are mean and s.d. of three independent experiments.
Extended Data Figure 3 POLR2Aloss cells are highly sensitive to POLR2A inhibition.
a, Cell proliferation of POLR2Aneutral and POLR2Aloss cells treated with actinomycin D. b, Knockdown efficiency of POLR2A-specific shRNAs in HCT116, SW480, SW837 and SNU283 cells. shNT denotes non-targeting control shRNA. c, Effect of POLR2A knockdown on the proliferation of four colorectal cancer cell lines. Cells expressing GFP and control or POLR2A-specific shRNAs were sorted and mixed with control GFP-negative cells (1:1) and the GFP-positive cells were quantified at passages 2, 4 and 6. **P < 0.01; ns, not significant. d, Protein levels of POLR2A in HCT116 and SNU283 cells expressing Dox-inducible POLR2A shRNAs (1.0 μg ml−1 Dox). e, Cell proliferation of HCT116 and SNU283 cells expressing Dox-inducible POLR2A shRNA in the presence of 300 ng ml−1 Dox. **P < 0.01. f, g, Cell cycle profiles (f) and apoptosis (g) of control or POLR2A shRNA-expressing HCT116 and SNU283 cells. **P < 0.01. Data are mean and s.d. of three independent experiments.
Extended Data Figure 4 Ectopic expression of POLR2A restores the resistance of POLR2Aloss cells to α-amanitin treatment.
a, Protein levels of POLR2A in SNU283 and SW837 cells expressing increasing amounts of exogenous POLR2A. b, Crystal violet staining of SNU283 and SW837 cells treated with α-amanitin after transfection with increasing amounts of POLR2A expression vector DNA.
Extended Data Figure 5 Mono-allelic knockout of POLR2A sensitizes HCT116 cells to POLR2A inhibition.
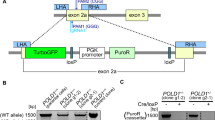
a, Schematic illustration of the Cas9/sgRNA-targeting sites in the POLR2A gene. Two single-guide RNA (sgRNA)-targeting sequences are shown and the protospacer-adjacent motif (PAM) sequences are highlighted in red. b, Efficiency of the Cas9-mediated cleavage of POLR2A in HCT116 cells measured by the Surveyor assay. c, Sequences of mutant POLR2A alleles in the cell colonies 14 and 5. PAM sequences are highlighted in red. Small deletions in the targeted region led to open reading frame shift, producing only a short stretch of the amino-terminal peptide without any functional domains of POLR2A. d, Protein levels of POLR2A in POLR2Aneutral and POLR2Aloss HCT116 cells. e, Growth curves of POLR2Aneutral and POLR2Aloss HCT116 cells. f, Relative proliferation of POLR2Aneutral and POLR2Aloss cells treated with actinomycin D. g, Effect of POLR2A knockdown on the POLR2Aneutral and POLR2Aloss HCT116 cells. Experiments were performed as described in Extended Data Fig. 3c. **P < 0.01. h, Dox-induced partial suppression of POLR2A inhibited the growth of POLR2Aloss HCT116 cells, but not of parental POLR2Aneutral HCT116 cells. Data are mean and s.d. of three independent experiments.
Extended Data Figure 6 Sensitivity of POLR2Aloss cells to POLR2A inhibition is independent of p53.
a, Schematic illustration of the Cas9/sgRNA-targeting sites in the TP53 gene. Two sgRNA-targeting sequences are shown and the PAM sequences are highlighted in red. b, Efficiency of the Cas9-mediated cleavage of TP53 in HCT116 cells measured by Surveyor assay. c, Protein levels of POLR2A and p53 in a panel of isogenic HCT116 cells. d, Growth curves of POLR2Aneutral and POLR2Aloss xhCRC cells. e, Growth curves of POLR2Aneutral and POLR2Aloss HCT116 cells. f, g, Crystal staining images (f) and cell survival curves (g) of POLR2Aneutral and POLR2Aloss HCT116 cells treated with α-amanitin. h, Cell survival curves of POLR2Aneutral and POLR2Aloss HCT116 cells in response to the treatment of ama–HEA125. Data are mean and s.d. of three independent experiments.
Extended Data Figure 7 Dose-dependent suppression of POLR2A inhibits tumorigenesis in POLR2Aloss, but not POLR2Aneutral tumours.
a, Quantification of POLR2A mRNA expression levels in subcutaneously xenografted HCT116 and SNU283 tumours expressing control or POLR2A shRNA (n = 5 mice per group). **P < 0.01. Data are mean and s.d. b, Immunohistochemical staining of the aforementioned xenograft tumours. HE, haematoxylin and eosin. c, Cells positive for Ki67 (cell proliferation) or cleaved caspase-3 (apoptosis) per field and POLR2A expression in b were quantified. **P < 0.01. n = 10 fields. Data are mean and s.d. d, Gross tumour images of xenograft tumours derived from subcutaneously implanted POLR2Aneutral and POLR2Aloss HCT116 cells (1 × 106 cells injected). Both cell lines express control or Dox-inducible POLR2A shRNAs. After the initial establishment of tumours (100 mm3), mice were treated with Dox (0.5, 1 and 2 μg ml−1) in drinking water. n = 5 mice per group. e, Quantification of tumour sizes as shown in d. Data are mean and s.d. f, Representative bioluminescent images of orthotopically implanted HCT116 tumours expressing Dox-inducible control or POLR2A shRNA after Dox treatment.
Extended Data Figure 8 Suppression of POLR2A with DOPC-encapsulated POLR2A siRNA inhibits the growth of POLR2Aloss tumours.
a, Protein levels of POLR2A after transfection of control siRNA or POLR2A siRNAs (shPol2-1 and shPol2-2) in HCT116 cells. b, Schematic illustration of orthotopic injection of HCT116 cells (1 × 106 cells) followed by siRNA–DOPC nanoliposome treatment. c–f, Representative bioluminescent images (c, e) and tumour growth curves (d, f) of orthotopic xenograft tumours derived from POLR2Aneutral and POLR2Aloss HCT116 cells that received intraperitoneal injections of control (1,000 μg kg−1) or POLR2A siRNAs (125, 250, 500 and 1,000 μg kg−1) twice weekly. n = 10 mice per group. Error bars, s.e.m. g, h, Representative protein levels of POLR2A in xenograft tumours after control or POLR2A siRNA treatment.
Extended Data Figure 9 Suppression of POLR2A selectively inhibits the POLR2Aloss tumour growth.
a, Immunohistochemical staining of xenografted xhCRC tumours. b, c, Tumour weights of orthotopically implanted HCT116 (b) and xhCRC (c) tumours. n = 10 mice per group. Data are mean and s.d. d, e, Body weights (d) and liver enzymes (e) including alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase in peripheral blood. Data are mean and s.d. n = 5 mice.
Extended Data Figure 10 Suppression of POLR2A by ama–HEA125 inhibits the growth of POLR2Aloss tumours.
a, d, g, Protein levels of POLR2A in HCT116 (a), SW480 (d) or SW837 (g) cells. These cell lines are POLR2Aneutral, POLR2Aloss or POLR2A-restored. b, c, e, f, h, i, Representative bioluminescent images (b, e, h) and tumour growth curves (c, f, i) of orthotopic xenograft tumours derived from the corresponding cells as indicated. All of them received dual intraperitoneal injections of anti-EpCAM antibody (3.6 mg kg−1) or ama–HEA125 antibody–drug conjugate (10 and 90 μg kg−1, corresponding to 0.4 and 3.6 mg IgG kg−1). n = 10 mice per group. Error bars, s.e.m.
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1-2 and original Western blotting images. (PDF 1903 kb)
Rights and permissions
About this article

Cite this article
Liu, Y., Zhang, X., Han, C. et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520, 697–701 (2015). https://doi.org/10.1038/nature14418
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14418
This article is cited by
-
Crosstalk between colorectal CSCs and immune cells in tumorigenesis, and strategies for targeting colorectal CSCs
Experimental Hematology & Oncology (2024)
-
Exploring the next generation of antibody–drug conjugates
Nature Reviews Clinical Oncology (2024)
-
Improved prediction of prognosis and therapy response for lung adenocarcinoma after identification of DNA-directed RNA polymerase-associated lncRNAs
Journal of Cancer Research and Clinical Oncology (2023)
-
Sestrin2 reduces cancer stemness via Wnt/β-catenin signaling in colorectal cancer
Cancer Cell International (2022)
-
Hypoxia-induced circWSB1 promotes breast cancer progression through destabilizing p53 by interacting with USP10
Molecular Cancer (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
