
Abstract
Given the importance of Africa to studies of human origins and disease susceptibility, detailed characterization of African genetic diversity is needed. The African Genome Variation Project provides a resource with which to design, implement and interpret genomic studies in sub-Saharan Africa and worldwide. The African Genome Variation Project represents dense genotypes from 1,481 individuals and whole-genome sequences from 320 individuals across sub-Saharan Africa. Using this resource, we find novel evidence of complex, regionally distinct hunter-gatherer and Eurasian admixture across sub-Saharan Africa. We identify new loci under selection, including loci related to malaria susceptibility and hypertension. We show that modern imputation panels (sets of reference genotypes from which unobserved or missing genotypes in study sets can be inferred) can identify association signals at highly differentiated loci across populations in sub-Saharan Africa. Using whole-genome sequencing, we demonstrate further improvements in imputation accuracy, strengthening the case for large-scale sequencing efforts of diverse African haplotypes. Finally, we present an efficient genotype array design capturing common genetic variation in Africa.
Similar content being viewed by others

Main
Globally, human populations show structured genetic diversity as a result of geographical dispersion, selection and drift. Understanding this variation can provide insights into evolutionary processes that shape both human adaptation and variation in disease susceptibility1. Although the Hapmap Project2 and the 1000 Genomes Project3 have greatly enhanced our understanding of genetic variation globally, the characterization of African populations remains limited. Other efforts examining African genetic diversity have been limited by variant density and sample sizes in individual populations4, or have focused on isolated groups, such as hunter gatherers (HG)5,6, limiting relevance to more widespread populations across Africa.
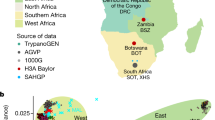
The African Genome Variation Project (AGVP) is an international collaboration that expands on these efforts by systematically assessing genetic diversity among 1,481 individuals from 18 ethno-linguistic groups from sub-Saharan Africa (SSA) (Fig. 1 and Supplementary Methods Tables 1 and 2) with the HumanOmni2.5M genotyping array and whole-genome sequences (WGS) from 320 individuals (Supplementary Methods Table 2). Importantly, the AGVP has evolved to help develop local resources for public health and genomic research, including strengthening research capacity, training, and collaboration across the region. We envisage that data from this project will provide a global resource for researchers, as well as facilitate genetic studies in Africa7.

a, 18 African populations studied in the AGVP including 2 populations from the 1000 Genomes Project. (The term ‘Ethiopia’ encompasses the Oromo, Amhara and Somali ethno-linguistic groups.) b, c, ADMIXTURE analysis of these 18 populations alone (n = 1,481) (b) and in a global context (n = 3,904) (c). Each colour represents a different ancestral cluster, with clusters 2–6 represented along the y-axis in b and clusters 2–18 represented in c. K = 6 and K = 18 were the most likely clusters on ADMIXTURE analysis. ADMIXTURE analysis suggests substructure between North, East, West and South Africa. Studying these populations in the context of Eurasian and African HG populations suggest extensive Eurasian and HG admixture across Africa.
Population structure in SSA
On examining ∼2.2 million variants, we found modest differentiation among SSA populations (mean pairwise FST 0.019) (Supplementary Methods and Supplementary Table 1). Differentiation among the Niger-Congo language groups—the predominant linguistic grouping across Africa was noted to be modest (mean pairwise FST 0.009) (Supplementary Table 1), providing evidence for the ‘Bantu expansion’—a recent population expansion and movement throughout SSA originating in West Africa around 3,000 to 5,000 years ago8.
We identified 29.8 million single-nucleotide polymorphisms (SNPs) from Ethiopian, Zulu and Bagandan WGS (Extended Data Fig. 1 and Supplementary Methods). A substantial proportion of unshared (11%–23%) and novel (16%–24%) variants were observed, with the highest proportion among Ethiopian populations (Extended Data Fig. 1). The high proportion of unshared variation among populations recapitulates the need for large-scale sequencing across Africa, including among genetically divergent populations.
We used principal component analysis to explore relationships among AGVP populations (Extended Data Figs 2, 3, 4, 5, Supplementary Figs 1 and 2). PC1 appeared to represent a cline extending from West and East African populations towards Ethiopian populations, possibly suggesting Eurasian gene flow, while PC2 separated West African and South/East African populations (Extended Data Fig. 2). Inclusion of the 1000 Genomes Project, North African and Khoe-San (Khoisan) populations in principal component analysis (Extended Data Figs 3, 4, 5, and Supplementary Figs 1 and 2) suggested possible HG ancestry among southern Niger-Congo groups—highlighted by clustering towards the Khoe-San, in addition to confirming a cline towards Eurasian populations. ‘Unsupervised’ (that is, without including known information on individual ancestry) ADMIXTURE9 (https://www.genetics.ucla.edu/software/admixture/) analysis including the 1000 Genomes Project and Human Origins data sets (Fig. 1), also supported evidence for substantial Eurasian and HG ancestry in SSA (Fig. 1 and Extended Data Fig. 6).
To assess the effect of gene flow on population differentiation in SSA, we masked Eurasian ancestry across the genome (Supplementary Methods and Supplementary Note 6). This markedly reduced population differentiation, as measured by a decline in mean pairwise FST from 0.021 to 0.015 (Supplementary Note 6), suggests that Eurasian ancestry has a substantial impact on differentiation among SSA populations. We speculate that residual differentiation between Ethiopian and other SSA populations after masking Eurasian ancestry (pairwise FST = 0.027) may be a remnant of East African diversity pre-dating the Bantu expansion10.
Population admixture in SSA
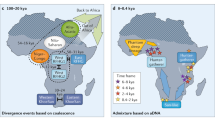
Formal tests for admixture (the three population test or f3 statistic)11, confirmed widespread Eurasian and HG admixture in SSA (Supplementary Tables 2 and 3). Quantification of admixture (Supplementary Table 4, Supplementary Methods and Supplementary Notes 3 and 4) indicated substantial Eurasian ancestry in many African populations (ranging from 0% to 50%), with the greatest proportion in East Africa (Fig. 2 and Supplementary Table 4). Similarly, HG admixture ranged from 0% to 23%, being greatest among Zulu and Sotho (Fig. 2 and Supplementary Table 5).

The proportion and distribution of Eurasian and HG admixture among different populations across Africa, with approximate dating of admixture using MALDER (code was provided by J. Pickrell; see Supplementary Information).
We found evidence for historically complex and regionally distinct admixture with multiple HG and Eurasian populations across SSA (Fig. 2 and Supplementary Note 5). Specifically, ancient Eurasian admixture was observed in central West African populations (Yoruba; ∼7,500–10,500 years ago), old admixture among Ethiopian populations (∼2,400–3,200 years ago) consistent with previous reports10,12, and more recent complex admixture in some East African populations (∼150–1,500 years ago) (Fig. 2, Extended Data Fig. 7 and Supplementary Note 5). Our finding of ancient Eurasian admixture corroborates findings of non-zero Neanderthal ancestry in Yoruba, which is likely to have been introduced through Eurasian admixture and back migration, possibly facilitated by greening of the Sahara desert during this period13,14.
We also find evidence for complex and regionally distinct HG admixture across SSA (Fig. 2, Extended Data Figs 7 and Supplementary Note 5), with ancient gene flow (∼9,000 years ago) among Igbo and more recent admixture in East and South Africa (multiple events ranging from 100 years ago to 3,000 years ago), broadly consistent with historical movements reflecting the Bantu expansion. An exploration of the likeliest sources of admixture in our data suggested that HG admixture in Igbo was most closely represented by modern day Khoe-San populations rather than by rainforest HG populations (Supplementary Note 5). Given limited archaeological and linguistic evidence for the presence of Khoe-San populations in West Africa, this extant HG admixture might represent ancient populations, consistent with the presence of mass HG graves from the early Holocene period comprising skeletons with distinct morphological features15, and with evidence of HG rock art dating to this period in the western Sahara16,17. In East Africa, our analyses suggested that Mbuti rainforest HG populations most closely represented ancient HG mixing populations (Supplementary Note 5), with admixture dating to ∼3,000 years ago, suggesting that HG ancestry here is likely to be older than previously reported18. The primary source of HG admixture in Zulu and Sotho populations was from Khoe-San populations (Fig. 2 and Supplementary Note 5), consistent with linguistic assimilation of click consonants among these populations.
Positive selection in SSA
We examined highly differentiated SNPs between European and African populations, as well as among African populations to gain insights into loci that may have undergone selection in response to local adaptive forces (Supplementary Methods). To account for confounding due to Eurasian admixture, we also conducted analyses after masking Eurasian ancestry (Supplementary Methods and Supplementary Note 6).
On examining locus-specific Europe–Africa differentiation, enrichment of loci known to be under positive selection was observed among the most differentiated sites (P = 1.4 × 10−31). Furthermore, there was statistically significant enrichment for gene variants among these, indicating that this differentiation is unlikely to have arisen purely from random drift (P = 0.0002). Additionally, we found no evidence for background selection as the primary driver of differentiation among these loci (Supplementary Note 7).
In addition to genes known to be under positive selection (for example, SLC24A5, SLC45A2 and OCA219,20, LARGE21 and CYP3A4/5) (Supplementary Fig. 3), we found evidence of differentiation in novel gene regions, including one implicated in malaria (for chemokine receptor 1, CR1) (Extended Data Fig. 8). CR1 carries the Knops blood group antigens and has previously been implicated in malaria susceptibility22 and severity23, with evidence suggesting positive selection in malaria-endemic regions24 (Extended Data Fig. 8). We also identified highly differentiated variants within genes involved in osmoregulation (ATP1A1 and AQP2) (Extended Data Fig. 8). Deregulation of AQP2 expression and loss-of-function mutations in ATP1A1 have been associated with essential and secondary hypertension, respectively25,26. Climatic adaptive changes in these gene regions could potentially provide a biological basis for the high burden of hypertension and differences in salt sensitivity observed in SSA27.
In contrast, overall differentiation among African populations was modest (maximum masked FST = 0.19) (Supplementary Fig. 4) and only 56/1,237 sites remained in the tail distribution after masking (Supplementary Methods, Supplementary Table 6). This suggests that a large proportion of differentiation observed among African populations could be due to Eurasian admixture, rather than adaptation to selective forces (Supplementary Note 6). Genes known to be under selection were notably enriched among the most differentiated loci after masking of Eurasian ancestry (P = 2.3 × 10−16). Among the 56 loci robust to Eurasian ancestry masking (Supplementary Table 6), we identified several loci known to be under selection (Extended Data Fig. 8), including a highly differentiated variant (rs1378940) in the CSK gene region implicated in hypertension in genome-wide association studies (GWAS)28. The major allele of rs1378940 among Africans was in complete linkage disequilibrium with the risk allele of the GWAS SNP rs1378942 (ref. 29), with the frequency of this allele highly correlated with latitude (r = −0.67), providing support for local adaptation in response to temperature as a possible mechanism for hypertension (Supplementary Fig. 5)30,31.
Comparing populations residing in endemic and non-endemic infectious disease regions (Supplementary Methods), we identified several loci associated with infectious disease susceptibility and severity. As well as the known sickle-cell locus related to malaria, this approach identified additional signals for genes potentially under selection, including the PKLR region32, RUNX333, the haptoglobin locus, CD16334, IL1035,36, CFH, and the CD28-ICOS-CLTA4 locus (Supplementary Table 7 and Extended Data Fig. 8)37. Similar comparisons for Lassa fever identified the known LARGE gene, as well as candidates associated with viral entry and immune response, including in the Histocompatibility Leukocyte Antigen region, DC-SIGN/DC-SIGNR38 (also known as CD209/CLEC4M), RNASEL, CXCR6, IFIH139 and OAS2/3 regions (Supplementary Table 7). For trypanosomiasis, we identified APOL140, as well as several loci implicated in immune response and binding to trypanosoma, including FAS, FASLG41,42, IL23R43, SIGLEC6 and SIGLEC12 (Supplementary Table 7)44. For trachoma, we identified signals in ABCA1 and CXCR6, which may be important for the growth of the parasite and host immune response, respectively (Supplementary Table 7)45,46.
Designing medical genetics studies in Africa
To inform the design of genomic studies in Africa, we addressed the following questions: (1) How well do current genotype arrays perform in African populations using existing reference panels for imputation? (2) Can these genotype arrays and reference panels identify and fine-map association signals in populations across Africa? (3) Can we improve imputation accuracy in African populations using a new African reference panel? and (4) What are the most cost-effective designs for large-scale GWAS in Africa?
The 1000 Genomes Project phase I integrated panel provided reasonably accurate imputation into the Illumina Omni 2.5M array in all populations (Supplementary Note 10). However, imputation accuracy was lower among Sotho, Zulu and Afro-Asiatic populations, possibly reflecting poor representation of some African haplotypes (including Khoe-San haplotypes) within the 1000 Genomes Project panel. These findings suggest that improvements in imputation accuracy across diverse population groups may require larger and more diverse reference panels.
We assessed the reproducibility and potential for fine-mapping association signals within Africa and globally at several disease susceptibility loci (Supplementary Methods, Supplementary Table 8 and Extended Data Fig. 9). Current genotype arrays and imputation panels allowed for identification of relevant association signals at most loci across populations in SSA, demonstrating that association signals are reproducible across populations in SSA (Extended Data Fig. 9 and Supplementary Figs 7–18). African populations are likely to provide better fine-mapping resolution around the causal locus (Supplementary Table 8). We highlight one example here: the sickle-cell anaemia locus (HBB)47, which is under positive selection owing to the protection the sickle cells confer against severe malaria. This locus showed marked heterogeneity in association signals across populations, reflecting different linkage disequilibrium patterns and allele frequencies among populations in SSA (Supplementary Figs 9 and 10). This pattern is probably the result of independent selection sweeps at this locus in different parts of Africa, leading to differences in hitchhiking rare haplotypes that attained high frequencies among different populations48. This suggests that these signatures are recent and occurred during or after the Bantu expansion, consistent with the hypothesis that the advent of agriculture and increased malaria transmission may have resulted in increased selection pressure49. However, in contrast to previous reports47, we show that association signals even at such highly differentiated loci can be captured with dense genotype data using existing reference panels for imputation, despite individual population groups not being fully represented in these. This suggests that, instead of large-scale population-specific sequencing across Africa, what is needed is a broad sequencing approach, targeted at capturing widespread haplotype diversity.
To assess the utility of a larger and more diverse African reference panel for imputation, we generated a panel integrating the 1000 Genomes Project phase I and AGVP WGS panels (Supplementary Methods and Supplementary Note 9). Using this integrated panel, we observed marked improvements in imputation accuracy across the whole range of the allele frequency spectrum in specific populations poorly represented by the 1000 Genomes Project panel (Fig. 3 and Supplementary Note 11). These findings suggest that even common haplotypes in some SSA populations may not be sufficiently captured by existing panels, limiting our power to examine associations of common variants with disease. Importantly, given the specificity of the improvement in imputation accuracy, we infer that targeted sequencing of divergent populations representing a broad spectrum of haplotypes across Africa, including HG and North/East African haplotypes, rather than widespread population sequencing is likely to provide a more efficient strategy to improve imputation accuracy and a practicable GWAS framework in Africa.

The substantial improvement in imputation accuracy in some populations (Sotho), compared to minimal improvement in others (Igbo) with the addition of the AGVP WGS reference panel to the 1000 Genomes Project phase I reference panel (‘merged’) suggests poor representation of some haplotypes (for example, Khoe-San haplotypes in Sotho) in the 1000 Genomes Project reference panel alone (‘1000’). r2 is the correlation coefficient, representing the correlation between imputed and genotyped data, on masking each genotyped variant during imputation. MAF, minor allele frequency.
We compared the utility of existing chip designs (2.5M Illumina) and ultralow-coverage WGS designs (0.5×, 1×, 2× coverage) to determine the optimal design for African GWAS. Sensitivity for common variation was >90% at all sequencing depths (Supplementary Note 12). Examining the effective sample size for a fixed budget50, we found the effective sample size was greater for all ultralow-coverage WGS and chip array designs compared with 4× WGS. When computational costs were accounted for (Supplementary Note 12), the HumanOmni2.5M array provided the greatest effective sample size supporting the development and large-scale use of efficient genotype arrays in Africa, where these have been underutilized.
We therefore sought to evaluate a potential chip design to tag common variation across a wider range of African populations (Supplementary Note 13). Importantly, we show that an array with one million genetic variants could capture >80% of common variation (minor allele frequency >5%) across the genome (Extended Data Fig. 10). These analyses suggest that designing a pan-African genotype array to effectively capture common genetic variation across Africa is feasible, and could greatly facilitate large-scale genomic studies in Africa.
Discussion
The marked haplotype diversity within Africa has important implications for the design of large-scale medical genomics studies across the region, as well as studies of population history and evolution. In this context, the AGVP is a resource that will facilitate a broad range of genomic studies in Africa and globally.
Although Africa is the most genetically diverse region in the world, we provide evidence for relatively modest differentiation among populations representing the major sub-populations in SSA, consistent with recent population movement and expansion across the region beginning around 5,000 years ago—the Bantu expansion8. Although the history of the Bantu expansion is probably complex, assessments of population admixture can provide new insights. We note historically complex and regionally distinct admixture with multiple HG and Eurasian populations across SSA, including ancient HG and Eurasian ancestry in West and East Africa and more recent complex HG admixture in South Africa. As well as explaining genetic differentiation among modern populations in SSA, these admixture patterns provide genetic evidence for early back-to-Africa migrations, the possible existence of extant HG populations in western Africa—compatible with archaeological evidence15, and patterns of gene flow consistent with the Bantu expansion, including genetic assimilation of populations resident across the region.
This admixture also has important implications for the assessment of differentiation and positive selection in Africa. Accounting for these elements, we have identified loci under positive selection that are linked with hypertension, malaria, and other pathogens. This provides a proof-of-concept for the ability of geographically widespread genetic data within Africa to identify loci under selection related to diverse environments.
Our evidence for the broad transferability of genetic association signals and their statistical refinement has important implications for medical genetic research in Africa. Importantly, we highlight that such studies are feasible and can be enabled through the development of more efficient genotype arrays and diverse WGS reference panels for accurate imputation of common variation. In this context, we describe a framework for a new pan-African genotype array that could directly facilitate large-scale genomic studies in Africa.
A critical next step is the large-scale deep sequencing of multiple and diverse populations across Africa, which should be integrated with ancient DNA data. This would enable us to identify and understand signals of ancient admixture, patterns of historical population movements, and to provide a comprehensive resource for medical genomic studies in Africa.
References
Botigué, L. R. et al. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe. Proc. Natl Acad. Sci. USA 110, 11791–11796 (2013)
The International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003)
The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012)
Tishkoff, S. A. et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009)
Schlebusch, C. M. et al. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science 338, 374–379 (2012)
Jarvis, J. P. et al. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet. 8, e1002641 (2012)
The H3Africa Consortium. Enabling the genomic revolution in Africa. Science 344, 1346–1348 (2014)
de Filippo, C., Bostoen, K., Stoneking, M. & Pakendorf, B. Bringing together linguistic and genetic evidence to test the Bantu expansion. Proc. R. Soc. Lond. B 279, 3256–3263 (2012)
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009)
Pagani, L. et al. Ethiopian genetic diversity reveals linguistic stratification and complex influences on the Ethiopian gene pool. Am. J. Hum. Genet. 91, 83–96 (2012)
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012)
Pickrell, J. K. et al. Ancient west Eurasian ancestry in southern and eastern Africa. Proc. Natl Acad. Sci. USA 111, 2632–2637 (2014)
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014)
Kuper, R. & Kropelin, S. Climate-controlled Holocene occupation in the Sahara: motor of Africa's evolution. Science 313, 803–807 (2006)
Sereno, P. C. et al. Lakeside cemeteries in the Sahara: 5000 years of holocene population and environmental change. PLoS ONE 3, e2995 (2008)
The Bradshaw Foundation. The Origin of the Prehistoric Rock Art Artistshttp://www.bradshawfoundation.com/giraffe/artists.php (2014)
Tilman, L.-E. Rock art in African Highlands, Ennedi Highlands, Chad—Artists and Herders in a Lifeworld on the Margins. In Atlas of Cultural and Environmental Change in Arid Africa http://www.academia.edu/1580718/Rock_art_in_African_Highlands_Ennedi_Highlands_Chad_-_Artists_and_Herders_in_a_Lifeworld_on_the_Margins (Heinrich Barth Institute, 2007)
Patin, E. et al. The impact of agricultural emergence on the genetic history of African rainforest hunter-gatherers and agriculturalists. Nature Commun. 5, 3163, http://dx.doi.org/10.1038/ncomms4163 (2014)
Norton, H. L. et al. Genetic evidence for the convergent evolution of light skin in Europeans and East Asians. Mol. Biol. Evol. 24, 710–722 (2007)
Edwards, M. et al. Association of the OCA2 polymorphism His615Arg with melanin content in east Asian populations: further evidence of convergent evolution of skin pigmentation. PLoS Genet. 6, e1000867 (2010)
Andersen, K. G. et al. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Phil. Trans. R. Soc. Lond. B 367, 868–877 (2012)
Eid, N. A. et al. Candidate malaria susceptibility/protective SNPs in hospital and population-based studies: the effect of sub-structuring. Malar. J. 9, 119 (2010)
Panda, A. K. et al. Complement receptor 1 variants confer protection from severe malaria in Odisha, India. PLoS ONE 7, e49420 (2012)
Kosoy, R. et al. Evidence for malaria selection of a CR1 haplotype in Sardinia. Genes Immun. 12, 582–588 (2011)
Beuschlein, F. et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nature Genet. 45, 440–444, http://dx.doi.org/10.1038/ng.2550 (2013)
Graffe, C. C., Bech, J. N., Lauridsen, T. G., Vase, H. & Pedersen, E. B. Abnormal increase in urinary aquaporin-2 excretion in response to hypertonic saline in essential hypertension. BMC Nephrol. 13, 15 (2012)
Young, J. H. et al. Differential susceptibility to hypertension is due to selection during the out-of-Africa expansion. PLoS Genet. 1, e82 (2005)
Voight, B. F., Kudaravalli, S., Wen, X. & Pritchard, J. K. A map of recent positive selection in the human genome. PLoS Biol. 4, e72 (2006)
Tabara, Y. et al. Common variants in the ATP2B1 gene are associated with susceptibility to hypertension: the Japanese Millennium Genome Project. Hypertension 56, 973–980 (2010)
Hong, K. W. et al. Genetic variations in ATP2B1, CSK, ARSG and CSMD1 loci are related to blood pressure and/or hypertension in two Korean cohorts. J. Hum. Hypertens. 24, 367–372 (2010)
Levy, D. et al. Genome-wide association study of blood pressure and hypertension. Nature Genet. 41, 677–687 (2009)
Machado, P. et al. Malaria: looking for selection signatures in the human PKLR gene region. Br. J. Haematol. 149, 775–784 (2010)
Band, G. et al. Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet. 9, e1003509 (2013)
Kusi, K. A. et al. Levels of soluble CD163 and severity of malaria in children in Ghana. Clin. Vaccine Immunol. 15, 1456–1460 (2008)
Zhang, G. et al. Interleukin-10 (IL-10) polymorphisms are associated with IL-10 production and clinical malaria in young children. Infect. Immun. 80, 2316–2322 (2012)
Wilson, J. N. et al. Analysis of IL10 haplotypic associations with severe malaria. Genes Immun. 6, 462–466 (2005)
Jacobs, T., Graefe, S. E., Niknafs, S., Gaworski, I. & Fleischer, B. Murine malaria is exacerbated by CTLA-4 blockade. J. Immunol. 169, 2323–2329 (2002)
Shimojima, M., Stroher, U., Ebihara, H., Feldmann, H. & Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 86, 2067–2078 (2012)
Fumagalli, M. et al. Population genetics of IFIH1: ancient population structure, local selection, and implications for susceptibility to type 1 diabetes. Mol. Biol. Evol. 27, 2555–2566 (2010)
Ko, W. Y. et al. Identifying Darwinian selection acting on different human APOL1 variants among diverse African populations. Am. J. Hum. Genet. 93, 54–66 (2013)
Lopes, M. F. et al. Increased susceptibility of Fas ligand-deficient gld mice to Trypanosoma cruzi infection due to a Th2-biased host immune response. Eur. J. Immunol. 29, 81–89 (1999)
Martins, G. A. et al. Fas-FasL interaction modulates nitric oxide production in Trypanosoma cruzi-infected mice. Immunology 103, 122–129 (2001)
Ribeiro, C. M. et al. Trypanosomiasis-induced Th17-like immune responses in carp. PLoS ONE 5, e13012 (2010)
Crocker, P. R., Paulson, J. C. & Varki, A. Siglecs and their roles in the immune system. Nature Rev. Immunol. 7, 255–266 (2007)
Cox, J. V., Naher, N., Abdelrahman, Y. M. & Belland, R. J. Host HDL biogenesis machinery is recruited to the inclusion of Chlamydia trachomatis-infected cells and regulates chlamydial growth. Cell. Microbiol. 14, 1497–1512 (2012)
Natividad, A. et al. Human conjunctival transcriptome analysis reveals the prominence of innate defense in Chlamydia trachomatis infection. Infect. Immun. 78, 4895–4911 (2010)
Jallow, M. et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nature Genet. 41, 657–665 (2009)
Teo, Y. Y. et al. Genome-wide comparisons of variation in linkage disequilibrium. Genome Res. 19, 1849–1860 (2009)
Hedrick, P. W. Population genetics of malaria resistance in humans. Heredity 107, 283–304 (2011)
Pasaniuc, B. et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nature Genet. 44, 631–635 (2012)
Acknowledgements
This project was funded in part by the Wellcome Trust (grant number WT077383/Z/05/Z), The Wellcome Trust Sanger Institute (grant number WT098051), the Bill and Melinda Gates Foundation, the Foundation for the National Institutes of Health (grant number 566), and the UK Medical Research Council (grant numbers G0901213-92157, G0801566, G0600718 and MR/K013491/1). We also acknowledge the National Institute for Health Research Cambridge Biomedical Research Centre and the Wellcome Trust Cambridge Centre for Global Health Research. We are very grateful to J. Pickrell for sharing human origins data and MALDER code, and for useful input on interpretations of these analyses. We also thank E. Garrison for his suggestions on using Genome-in-a-bottle sets (ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/variant_calls/NIST/README.NIST.v2.18.txt) for validation of whole-genome sequencing data. We also thank the African Partnership for Chronic Disease Research (APCDR) for providing a network to support this study as well as a repository for deposition of curated data. Sample collections from South Africa were funded by The South African Sugar Association, Servier South Africa and The Victor Daitz Foundation. The Kenyan samples were collected by D. Ngare of Moi University, Eldoret, Kenya, as part of the Africa America Diabetes Mellitus (AADM) study and the International HapMap project (D. Ngare, who is now deceased, was a great supporter of genomics in Africa, as exemplified by his leadership in engaging the Luhya and Maasai communities for the HapMap project). The Igbo samples were collected by J. Oli of the University of Nigeria, Enugu, Nigeria. The Ga-Adangbe samples were collected by the laboratories of A. Amoah of the University of Ghana, Accra, Ghana, and J. Acheampong of the University of Science and Technology, Kumasi, Ghana. Support for the AADM study is provided by the National Institute on Minority Health and Health Disparities, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the National Human Genome Research Institute (NHGRI). The Gambian samples were collected by M. Jallow and colleagues at the MRC Unit, The Gambia and form part of the MalariaGEN Consortial Resource. This research was supported in part by the Intramural Research Program of the Center for Research on Genomics and Global Health (CRGGH; grant number Z01HG200362) and by the MRC Centre for Genomics and Global Health. D.G. was funded by the Cambridge Commonwealth Scholarship. We thank the 1000 Genomes Project for sharing genotype data that were analysed as part of this project. We also thank all study participants who contributed to this study.
Author information
Authors and Affiliations
Contributions
Overall project coordination: D.G., C.P., M.S.S. (Project Chair), E.H.Y. and E.Z. coordinated the project. Analysis and writing: C.P. coordinated sample collation, genotyping, quality control and data generation for the study. J.A., T.C., D.G. and C.P. carried out quality control and curation of data. R.N. and Y.X. undertook quality control for MalariaGEN and Ethiopian population sets respectively. M.O.P. carried out quality control and bam (sequencing reads file format) improvement of sequence data at all depths. T.C. curated and generated all sequence data, and carried out comparisons with genotype array data and with higher coverage data. D.G. carried out the population structure and admixture analyses. A.C., D.G., S.K. and L.P. carried out analysis of positive selection and population differentiation. L.P. and I.T. carried out analysis of linkage disequilibrium decay. T.C., K.H. and I.T. carried out imputation-based analyses. T.C. developed an efficient tagging algorithm and carried out analysis for coverage of tagging variants for the design of the African genotype array. D.G. and F.T.-A. carried out fine mapping analyses. C.R., M.S.S., C.T.-S. and E.Z. critically appraised and commented on the manuscript. D.G., T.C., L.P. and M.S.S. prepared the manuscript and the Supplementary Information. C.P. and L.I. contributed to the writing of the Supplementary Information. All authors commented on the interpretation of results, and reviewed and approved the final manuscript. Management, fieldwork, laboratory analyses and coordination of contributing cohorts: K.B., M.J., K.K., D.K., K.R. and F.S.-J. (the Gambian cohorts—MalariaGEN); G.A., P.K., A.K., M.S.S. and J.S. (The General Population Cohort Study); A.M. and F.P. (the South African Zulu cohort); A.A., A.P.D., C.R. and F.T.-A. (the Kenyan, Ghanaian and Nigerian cohorts); A.C., S.N., M.R. and S.T. (the South African Sotho cohort); and E.B., N.B., R.E., E.M., T.O., L.P and C.T. (the Ethiopian cohort).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
The ADMIXTURE code is available at https://www.genetics.ucla.edu/software/admixture/download.html. The MALDER software is available from J. Pickrell (jkpickrell@nygenome.org). All other source code can be obtained by contacting D.G. (dg11@sanger.ac.uk). See Supplementary Methods for details.
Extended data figures and tables
Extended Data Figure 1 Allele sharing between sequenced populations in the AGVP.
a, The overlap of SNPs between 4×WGS data from Zulu, Ugandan and Ethiopian individuals (subsampled to 100 samples each). b, The overlap of novel variants (those not in the 1000 Genomes Project phase I integrated call set, ‘1000G’) between the three populations. c, d, The allele frequency spectra of variants in different portions of the Venn diagrams depicted in a and b, respectively. There appear to be a large proportion of unshared (private) variants in each population: between 10% and 23% of the total number of variants in a given population. The proportion of novel variants was high, with Ethiopia showing the greatest proportion of novel variation. Most of the novel variation appears to be unshared and rare.
Extended Data Figure 2 The first ten principal components for the African data set.
PC1 shows a cline among several African populations, most likely to represent Eurasian gene flow (n = 1,481). PC2 shows a clear separation between West and South/East Africa. Subsequent PCs show more detailed structure between, and within African populations.
Extended Data Figure 3 The first ten principal components for the global data set, including populations from the 1000 Genomes Project.
PC1 shows a cline among several African populations extending towards European populations, most likely to represent non-SSA gene flow (n = 2,864). PC2 shows a clear separation between European and Asian populations. Subsequent PCs show more detailed structure between populations globally, and within African populations. GBR, British in England and Scotland; ACB, African Caribbeans in Barbados; ASW, Americans of African ancestry in southwestern USA; CDX, Chinese Dai in Xishuangbanna, China; CEU, Utah residents with Northern and Western European ancestry; CHB, Han Chinese in Beijing, China; CHS, Southern Han Chinese; CLM, Colombians from Medellin, Colombia; FIN, Finnish in Finland; GIH, Gujarati Indian from Houston, Texas, USA; IBS, Iberian population in Spain; JPT, Japanese in Tokyo, Japan; KHV, Kinh in Ho Chi Minh City, Vietnam; MXL, Mexican ancestry from Los Angeles, USA; PEL, Peruvians from Lima, Peru; PUR, Puerto Ricans from Puerto Rico, and TSI, Toscani in Italy.
Extended Data Figure 4 The first ten principal components for the global extended data set, including populations from the 1000 Genomes Project, Human Genome Diversity Project, North African and Khoe-San population groups.
PC1 shows a cline among several African populations extending towards European populations, most likely to represent non-SSA gene flow (n = 3,202). PC2 shows a clear separation between European and Asian populations. Subsequent principal components show more detailed structure between populations globally, and within African populations.
Extended Data Figure 5 Projection of principal components to assess admixture among African populations.
a, The projection of principal components calculated on YRI and CEU from the 1000 Genomes Project onto the African populations. The AGVP populations are seen to fall on a cline between YRI and CEU, with Ethiopian populations closest to CEU. This is suggestive of Eurasian ancestry among these populations. b, The projection of principal components calculated on YRI and Ju/’hoansi onto the AGVP and other Khoe-San populations. The AGVP and Khoe-San populations are seen to fall on a cline between YRI and Ju/’hoansi, with Zulu and Sotho leading the cline among the AGVP populations. This is suggestive of HG gene flow among these populations.
Extended Data Figure 6 ADMIXTURE clustering analysis for AGVP samples combined with the 1000 Genomes Project, Human Genome Diversity Project, North African and Khoe-San samples.
Cluster K = 2 shows separation of European and African ancestry, with delineation of Asian and Khoe-San ancestry in cluster K = 4. Subsequent clusters show separation of East, West, North and South African ancestral components n = 3,202.
Extended Data Figure 7 Dating and source of admixture in the AGVP.
a, The time and most likely sources of admixture with means and 95% confidence intervals for different AGVP populations estimated with MALDER (see Supplementary Note 5). Circular markers with a line drawn around them represent high-probability events, while those with no line around them represent low-probability events. b, The time and most likely sources of admixture estimated with MALDER for the same populations using high-quality imputed data to improve resolution.
Extended Data Figure 8 Loci with marked allelic differentiation either globally or within Africa.
The derived and ancestral alleles are depicted in blue and red, respectively, for all loci. a, The global distribution of the non-synonymous variant rs17047661 at the CR1 locus implicated in malaria severity. This locus was noted to be among the most differentiated sites (in the top 0.1%) between Europe and Africa. b, The global distribution of the rs10216063 SNP at the AQP2 locus. The derived allele appears to be the major allele among European populations in contrast to African populations. c, The allele frequency distribution of rs10924081 at the ATP1A1 locus. Marked differentiation is observed globally, with the derived allele noted to be the major allele among European populations. d, The global distribution of the risk allele for the SNP rs1378940 in the CSK locus associated with hypertension. This locus was found to be within the top 0.1% of differentiated loci within Africa, and within the top 1% of differentiated loci globally. e, The allele frequency distribution of the rs3213419 SNP at the HP locus. f, The allele frequency distribution of the rs7313726 SNP at the CD163 locus. The HP and CD163 are among the top 0.1% of differentiated sites between malaria endemic and non-endemic regions in Africa.
Extended Data Figure 9 The global distribution of biologically relevant loci used for simulation of traits to examine reproducibility of signals across AGVP populations.
a, The frequency of the sickle-cell variant (rs334) in different regions globally. The blue portion of each pie chart represents the frequency of the causal allele A. b, The distribution of the SORT1 causal SNP rs12740374, with the derived allele T depicted in blue. c–f, The distributions of the APOL1 variant rs73885319, TCF7L2 variant rs7903146, the APOE variant rs429358 and the PRDM9 variant rs6889665, respectively.
Extended Data Figure 10 The coverage obtained across the genome for variants at different allele frequencies for a hypothetical African genotype array with one million tagging variants.
Different allele frequency bins are depicted in different colours. The lines show the coverage that can be achieved by imputation at different r2 thresholds. Coverage, here, is defined as the proportion of variants within an allele frequency captured above a pre-defined r2 threshold (along the x axis) after imputation. The solid lines represent the coverage obtained with one million variants selected using the hybrid tagging and imputation approach, while the broken lines represent the coverage obtained by using a simple pairwise tagging approach to capture one million tagging variants. The hybrid method improves the coverage obtained, particularly for common variation. Coverage for common variants (>5%) appears to be high at an r2 threshold of 0.8 and above, with >80% of these variants accurately imputed.
Supplementary information
Supplementary Information
This file contains Supplementary Methods. (PDF 17935 kb)
Supplementary Data
This file contains Supplementary Tables 1-8 and Supplementary Figures 1-18. (PDF 17582 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Gurdasani, D., Carstensen, T., Tekola-Ayele, F. et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 517, 327–332 (2015). https://doi.org/10.1038/nature13997
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13997
This article is cited by
-
Genetic drivers of heterogeneity in type 2 diabetes pathophysiology
Nature (2024)
-
Spatial and temporal diversity of positive selection on shared haplotypes at the PSCA locus among worldwide human populations
Heredity (2023)
-
Exome-based mutation screening in South African children with primary congenital glaucoma
Eye (2023)
-
The genetic legacy of the expansion of Bantu-speaking peoples in Africa
Nature (2023)
-
Africa-specific human genetic variation near CHD1L associates with HIV-1 load
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
