
Abstract
Human immunodeficiency virus (HIV)-1 is able to replicate in primary human macrophages without stimulating innate immunity despite reverse transcription of genomic RNA into double-stranded DNA, an activity that might be expected to trigger innate pattern recognition receptors. We reasoned that if correctly orchestrated HIV-1 uncoating and nuclear entry is important for evasion of innate sensors then manipulation of specific interactions between HIV-1 capsid and host factors that putatively regulate these processes should trigger pattern recognition receptors and stimulate type 1 interferon (IFN) secretion. Here we show that HIV-1 capsid mutants N74D and P90A, which are impaired for interaction with cofactors cleavage and polyadenylation specificity factor subunit 6 (CPSF6) and cyclophilins (Nup358 and CypA), respectively1,2, cannot replicate in primary human monocyte-derived macrophages because they trigger innate sensors leading to nuclear translocation of NF-κB and IRF3, the production of soluble type 1 IFN and induction of an antiviral state. Depletion of CPSF6 with short hairpin RNA expression allows wild-type virus to trigger innate sensors and IFN production. In each case, suppressed replication is rescued by IFN-receptor blockade, demonstrating a role for IFN in restriction. IFN production is dependent on viral reverse transcription but not integration, indicating that a viral reverse transcription product comprises the HIV-1 pathogen-associated molecular pattern. Finally, we show that we can pharmacologically induce wild-type HIV-1 infection to stimulate IFN secretion and an antiviral state using a non-immunosuppressive cyclosporine analogue. We conclude that HIV-1 has evolved to use CPSF6 and cyclophilins to cloak its replication, allowing evasion of innate immune sensors and induction of a cell-autonomous innate immune response in primary human macrophages.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Accession codes
Accessions
ArrayExpress
Protein Data Bank
Data deposits
Structural coordinates have been deposited under PDB accession code 4IPZ. Microarray data are available from the EBI Array Express repository under accession no. E-MTAB-1437.
Change history
20 November 2013
Figure 3 was corrected and changes were made to the Acknowledgements section.
References
Lee, K. et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7, 221–233 (2010)
Schaller, T. et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439 (2011)
Price, A. J. et al. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 8, e1002896 (2012)
Ambrose, Z. et al. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J. Virol. 86, 4708–4714 (2012)
Tsang, J. et al. HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS 23, 2255–2263 (2009)
Besnier, C., Takeuchi, Y. & Towers, G. Restriction of lentivirus in monkeys. Proc. Natl Acad. Sci. USA 99, 11920–11925 (2002)
Iyer, S. R., Yu, D., Biancotto, A., Margolis, L. B. & Wu, Y. Measurement of human immunodeficiency virus type 1 preintegration transcription by using Rev-dependent Rev-CEM cells reveals a sizable transcribing DNA population comparable to that from proviral templates. J. Virol. 83, 8662–8673 (2009)
Shi, J., Zhou, J., Shah, V. B., Aiken, C. & Whitby, K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85, 542–549 (2011)
Blair, W. S. et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 6, e1001220 (2010)
Dettwiler, S., Aringhieri, C., Cardinale, S., Keller, W. & Barabino, S. M. Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. J. Biol. Chem. 279, 35788–35797 (2004)
Ocwieja, K. E. et al. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 7, e1001313 (2011)
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013)
Gao, D. et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 (2013)
Dube, H. et al. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem. J. 441, 901–907 (2012)
Liu, J.-P., Ye, L., Wang, X., Li, J.-L. & Ho, W.-Z. Cyclosporin A inhibits hepatitis C virus replication and restores interferon-alpha expression in hepatocytes. Transpl. Inf. Dis. 13, 24–32 (2011)
Yan, N., Regalado-Magdos, A. D., Stiggelbout, B., Lee-Kirsch, M. A. & Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nature Immunol. 11, 1005–1013 (2010)
Manel, N. et al. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467, 214–217 (2010)
Mörner, A. et al. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J. Virol. 73, 2343–2349 (1999)
Zhang, F., Hatziioannou, T., Perez-Caballero, D., Derse, D. & Bieniasz, P. D. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology 353, 396–409 (2006)
Lee, K. et al. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J. Virol. 86, 3851–3860 (2012)
Ruepp, M. D., Schumperli, D. & Barabino, S. M. mRNA 3′ end processing and more–multiple functions of mammalian cleavage factor I-68. Wiley interdisciplinary reviews. RNA 2, 79–91 (2011)
Yang, Q., Gilmartin, G. M. & Doublie, S. The structure of human cleavage factor Im hints at functions beyond UGUA-specific RNA binding: a role in alternative polyadenylation and a potential link to 5′ capping and splicing. RNA Biol. 8, 748–753 (2011)
Hori, T. et al. A carboxy-terminally truncated human CPSF6 lacking residues encoded by exon 6 inhibits HIV-1 cDNA synthesis and promotes capsid disassembly. J. Virol. 87, 7726–7736 (2013)
Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L. & Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnol. 15, 871–875 (1997)
Naldini, L. et al. In vivo gene delivery and stable transduction of non-dividing cells by a lentiviral vector. Science 272, 263–267 (1996)
Nègre, D. et al. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 7, 1613–1623 (2000)
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378 (2003)
Lynn, D. J. et al. Curating the innate immunity interactome. BMC Syst. Biol. 4, 117 (2010)
Butler, S. L., Hansen, M. S. & Bushman, F. D. A quantitative assay for HIV DNA integration in vivo. Nature Med. 7, 631–634 (2001)
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009)
Collaborative Computational Project, number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 (1994)
Mikol, V., Kallen, J., Pflugl, G. & Walkinshaw, M. D. X-ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 Å resolution. J. Mol. Biol. 234, 1119–1130 (1993)
Acknowledgements
We are grateful to J. W. Chin, S. Goodbourn, K. Lee, O. Perisic and V. KewalRamani for reagents and advice. This work was funded by Wellcome Trust Senior Fellowship 090940 to G.J.T., the Medical Research Council, an MRC Confidence in Concept Award to G.J.T. and D.S. and the National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
J.R., C.P.T., A.J.F., D.L.S., M.N. and G.J.T. designed the study. J.R. performed all experiments in MDM, C.P.T. performed cGAMP and immunostimulatory RNA assays, A.J.F. performed CPSF6 experiments in HeLa cells. A.J.P. solved the structure of SmBz-CsC in complex with CypA and L.H. performed the TRIMCyp experiments. J.R., A.J.F., A.J.P., L.H., D.A.J., L.C.J., M.N. and G.J.T. analysed the data. A.J.F. and C.B. generated constructs and D.L.S. synthesized SmBz-CsA. J.R., M.N. and G.J.T. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.R., L.C.J., D.S. and G.J.T. are inventors on a patent claiming the anti-HIV activity of SmBz-CsA.
Extended data figures and tables
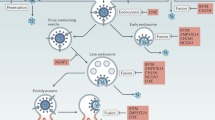
Extended Data Figure 1 A model for HIV-1 core behaviour and innate sensing.
a, Intact capsids recruit CypA and CPSF6 which direct the virus to the nucleus. CPSF6 interaction prevents premature DNA synthesis. Excess cytoplasmic DNA is degraded by TREX1. At the nuclear pore CPSF6 NLS-dependent dissociation from the virus allows reverse transcription to proceed. Reverse-transcribed DNA crosses the nuclear membrane and integrates. b, c, Disruption of CypA–CA interactions with either CA(P90A) mutation or cyclosporine treatment leads to detection of DNA reverse transcription product by cGAS initiating cGAMP production, STING activation, NF-κB/IRF3 nuclear localization, type I interferon secretion and initiation of an antiviral state. d, e, Disruption of CPSF6-CA interactions by N74D CA mutation, or depletion of CPSF6, leads to activation of a cryptic innate DNA sensor which also activates NF-κB/IRF3 nuclear localization. f, Disruption of CPSF6 engagement with the nuclear transport machinery by mutating its NLS prevents reverse transcription because the CPSF6 does not dissociate from the capsid at the nuclear pore. (g) PF74 mimics CPSF6 by inserting a phenyl ring into a CA pocket in the same position as CPSF6 and also prevents reverse transcription. Like CPSF6ΔNLS PF74 has no NLS and thus does not disengage from the core and therefore terminally prevents reverse transcription.
Extended Data Figure 2 HIV-1 mutants CA N74D and P90A, or WT HIV-1 on CPSF6 depletion, induce Type I IFN secretion in human macrophages that limits propagation.
a, MDM were infected with HIV-1 WT, CA N74D or CA(P90A) at low multiplicity. Cells were stained for Gag p24 at specific time points after infection and infected colonies counted. b, MDM transduced to express shRNA targeting CPSF6, or a scrambled control hairpin, were infected with wild-type NL4.3 (Ba-L Env) at low multiplicity. Cells were stained for Gag p24 at specific time points after infection and infected colonies counted. (a-b) P = 0.001, two-way ANOVA, for the effect of CA mutation or CPSF6 depletion. Data represent mean and nonlinear regression over time for 3 biological replicates. c, Individual experiments from two additional donors performed as experiments shown in Fig. 1a and b, and one additional donor performed as experiments in Fig. 2c and d. Each HIV-1 mutant is shown compared to wild-type virus data (WT) for comparison.
Extended Data Figure 3 Suppression of HIV-1 by type 1 interferon and rescue of infectivity with anti-IFN receptor (IFNAR2) antibody.
a, In order to demonstrate that IFN is able suppress wild type HIV-1 replication, MDM were pretreated with 1 ng ml−1 of recombinant IFN-β for 2 h then infected with HIV-1 WT NL4.3 (BaL-Env) infection. Cells were stained for Gag p24 at specific time points post infection (p.i). b, In order to determine how much IFN-α/β receptor (IFNAR2) neutralizing antibody is required to neutralize an IFN response, MDM were pretreated with varying concentrations of anti-IFNAR2 antibody for 2 h then stimulated with 1 ng ml−1 of recombinant IFN-β for 24 h. IP10 gene expression levels were measured by qRT–PCR and normalized to GAPDH. Results are expressed as fold change of expression over untreated cells. 1 μg ml−1 of IFNAR2 antibody effectively neutralized 1 ng ml−1 recombinant IFN-β, and this dose was used in subsequent experiments. c, MDM were infected with WT or WT and CA mutants at low multiplicity in the presence of anti-IFNAR2 antibody. Cells were stained for Gag at specific time points after infection and infected colonies counted (P values are given for two-way ANOVA for the effect of IFNAR blockade). Data represent mean and nonlinear regression of biological replicate experiments over time. d, Infectious titres of WT and CA mutant viruses were determined on MDM measured by assay of p24 positive cells 48 h post infection. Cells were infected in the presence of anti-IFNAR2 antibody or isotype control antibody (cAb). Titres are expressed as infectious units per nanogram of reverse transcriptase activity determined by ELISA. Mean ± s.e.m. of titre determined at 3 doses (technical replicates).
Extended Data Figure 4 Stimulation of gene expression by HIV-1 CA mutants in MDM.
a, b, Selected IFN-stimulated genes significantly upregulated by HIV-1 CA mutant infection, as well as by IFN-β and poly(I:C) measured at 24 h shown as fold change in expression (Stim/Control) measured by qRT–PCR and normalized to GAPDH mRNA levels. c, The same RNA samples as a, b were subjected to expression array and are presented in an expression matrix illustrating fold change in gene expression. d, Upregulation of gene expression (mean >2 fold in 2 independent biological replicates) after infection of MDM by HIV-1 wild-type and mutants HIV-1 CA N74D and HIV-1 CA(P90A) as shown. 24 h after infection (MOI 2), total RNA was isolated and subject to expression array, see methods. Results were subject to pathway analysis using the online bioinformatics tool- InnateDB (http://www.innatedb.com). Type 1 IFN signalling was the most significantly over-represented pathway with IFN-β, PolyIC and both HIV-1 mutants, but not WT virus, based on the Reactome database (http://www.reactome.org). The proportion of genes in each list that map to this pathway and the p-value following Benjamini-Hochberg correction for multiple biological replicates are indicated.
Extended Data Figure 5 Inhibition of HIV-1 with PF74 in MDM does not trigger IFN production.
a, Infection of human MDM with WT NL4.3 (Ba-L Env) in the presence or absence of 10 μM PF74. Cells were stained for p24 at specific time points after infection and infected colonies counted. b, Supernatants collected from MDMs in a were assayed for soluble IFN-β levels by ELISA. c, MDM were infected at low multiplicity with WT HIV-1 in the presence of PF74 and IFNAR2 antibody or isotype cAb. d, Measurement of HIV-1 late reverse transcription product (LRT) in MDM infected for 9 h with two concentrations of WT HIV-1, in the presence or absence of 10 µM PF74. Cells infected with boiled virus served as a negative control for DNA contamination. e, A sample parallel to those in d was used to determine the number of infected cells by staining for p24 48 h post infection. f, Measurement of HIV-1 LRT product in HeLa cells that express CPSF6ΔNLS or empty vector (EV), infected for 9 h with VSV-G pseudotyped HIV-1 GFP. g, A parallel sample was used to determine the number of infected cells by flow cytometry 48 h post infection. h, Infectious titres of WT and CA mutant viruses were determined on MDM in the presence and absence of SIVmac-VLPs encoding the SAMHD1 antagonist Vpx. Titres are expressed as infectious units per nanogram of reverse transcriptase activity determined by ELISA. Mean ± s.e.m. of titre determined at 3 doses. Experiments represent 3 independent biological replicates.
Extended Data Figure 6 HIV-1 P90A CA mutant infected MDM contained a benzonase and heat resistant component that activated an interferon sensitive promoter.
a, Immunoblot of extracts from L929 cells stably expressing Firefly Luciferase-driven by an interferon sensitive response element depleted of STING by siRNA or expressing control siRNA. b, L929 cells, control or STING depleted as in a, were treated with various concentrations of STING agonist cyclic GMP-AMP (cGAMP), and Luciferase activity was read after 16 h. c, MDM were infected with HIV-1 wild-type (WT) or mutant HIV-1 CA N74D or HIV-1 CA(P90A) for 18 h (MOI of 2), total cell extracts were isolated and were heat and benzonase treated and were applied to L929 cells as in a and Luciferase activity measured at 16 h (Mean of 4 ± s.e.m. of biological replicates). d, As c except RNA was extracted from the cell extracts and were transfected into 293T cells to measure IFN-β promoter driven Luciferase activity after 16 h. Sendai virus infection served as positive control for immuno-stimulatory RNA (Mean ± s.e.m. of biological replicates). c, * represents statistically significant difference between data sets (P < 0.05, t-test), NS represents non-significant differences.
Extended Data Figure 7 Nuclear translocation of NF-κB and IRF3 after HIV-1 capsid mutant infection in MDM.
a, Confocal immunofluorescence microscopy was used to quantify nuclear translocation of NF-κB Rel A (green) and IRF3 (red) as a consequence of activation. Nuclear:cytoplasmic ratios of immunostaining were measured at single cell level by quantitation of NF-κB or IRF3 signal intensities inside and outside the nucleus (blue DAPI) as described in methods. b, Data for 500 single cell measurements are shown for unstimulated MDM and MDM stimulated for 2 h with lipopolysaccharide (LPS) (100 ng ml−1), or infected with wild-type (WT) HIV-1 or N74D or P90A CA mutants. Red lines represent the mean of each data set. * represent statistically significant differences between data sets (P < 0.01, t-test), ns represents non-significant differences (P > 0.05, t-test).
Extended Data Figure 8 Inhibition of HIV-1 with cyclosporine or by TREX depletion triggers IFN production.
a, Infection of human MDM with WT NL4.3 (Ba-L Env) in the presence or absence of 5 μM cyclosporine. Cells were stained for p24 at specific time points after infection and infected colonies counted. b, Supernatants isolated from MDMs in a were assayed for soluble IFN-β levels by ELISA. c, MDM were infected at low multiplicity with WT HIV in the presence of cyclosporine and IFNAR2 antibody or isotype cAb. d, Infectious titre of WT HIV was determined on MDM in the presence of DMSO or cyclosporine at 48h post infection. The data are presented as infectious units (iu) per nanogram (ng) of reverse transcriptase (RT) measured by ELISA. e, To confirm that SmBz-CsA inhibits recruitment of cyclophilin A (CypA), but not Nup358 Cyp, to HIV-1 CA, we used the TRIMCyp restriction assay. HIV-1 GFP vector titer on CRFK cells expressing empty vector (EV), HA-tagged owl monkey TRIMCyp RBCC domain fused to human CypA (TRIMCypA) or human Nup358Cyp (TRIMNup358) in either DMSO, or 5 µM cyclosporine or 10 µM SmBz-CsA. Protein levels were measured by immunoblot detecting the HA tag with β-Actin as a loading control. In this assay both cyclosporine and SmBz-CsA inhibited CypA recruitment to CA and rescued VSV-G pseudotyped HIV-1 GFP infectivity from restriction by TRIMCypA. However, neither drug rescued HIV-1 infectivity from TRIMNup358, confirming cyclosporine specificity for CypA and not Nup358. f, TREX-1 expression was determined in MDM expressing TREX-1 specific shRNA or control shRNA by qRTPCR, normalized to GAPDH at the time of HIV-1 infection in g. g, Cells were stained for p24 at specific time points after WT HIV-1 infection of TREX-1 depleted and control shRNA expressing MDM and infected colonies counted. h, Supernatants isolated from MDM in g were assayed for soluble IFN-β by ELISA. i, Infection of TREX-1 depleted MDM with WT HIV-1 at low multiplicity in the presence of either IFNAR2 antibody or isotype cAb. Cells were stained for p24 at specific time points after infection and infected colonies counted. j, Hairpin transduced MDM were assayed for the production of soluble IFN-β levels before HIV-1 infection. Data are representative of 3 independent biological replicates. For HIV-1 replication assays in MDM data points and nonlinear regression lines over time are shown.
Extended Data Figure 9 HIV-1 CA mutants replicate without triggering IFN production in cell lines.
GHOST (a–c) or HeLa TZM bl (d–f) indicator cell lines were infected with WT NL4.3 (Ba-L Env) or NL4.3 (Ba-L Env) bearing CA mutations N74D or P90A at low multiplicity (0.04). Replication was monitored by GFP expression (GHOST) or staining LacZ positive cells (HeLa TZM bl). Both mutants replicated well, slightly behind wild-type virus. Replication in GHOST (b) or HeLa TZM bl (e) was performed in the presence of IFNAR2 antibody or isotype cAb. Neither antibody had any effect on WT or mutant HIV-1 replication in GHOST or HeLa TZM bl indicator cell lines. c, f, Induction of ISGs IP10 and IFIT1 expression were measured by quantitative RT PCR after high multiplicity infection (MOI 2) by WT or CA mutant HIV-1 on GHOST (c) or HeLa TZM bl (f). 10 ng ml−1 of IFN-β treatment acted as a positive control. ISG expression levels were normalized to GAPDH and are expressed as fold change in expression over unstimulated cells (Mean of 3 replicates ± s.e.m.). Neither WT or CA mutant HIV-1 induced ISG expression in either cell line. g, HeLa TZM bl were infected WT or CA G89V at low multiplicity (0.04). Replication was monitored by staining LacZ positive cells. h, HIV-1 CA G89V replication in HeLa TZM bl was defective and not rescued by anti-IFNAR2 or isotype control antibodies. All results are representative of 2 biological replicates.
Rights and permissions
About this article
Cite this article
Rasaiyaah, J., Tan, C., Fletcher, A. et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405 (2013). https://doi.org/10.1038/nature12769
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12769
This article is cited by
-
Evasion of cGAS and TRIM5 defines pandemic HIV
Nature Microbiology (2022)
-
Capsid adaptation defines pandemic HIV
Nature Microbiology (2022)
-
Vpx enhances innate immune responses independently of SAMHD1 during HIV-1 infection
Retrovirology (2021)
-
Rotten to the core: antivirals targeting the HIV-1 capsid core
Retrovirology (2021)
-
The HIV-1 capsid and reverse transcription
Retrovirology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
