
Abstract
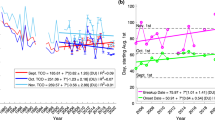
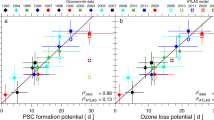
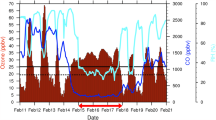
Chemical ozone destruction occurs over both polar regions in local winter–spring. In the Antarctic, essentially complete removal of lower-stratospheric ozone currently results in an ozone hole every year, whereas in the Arctic, ozone loss is highly variable and has until now been much more limited. Here we demonstrate that chemical ozone destruction over the Arctic in early 2011 was—for the first time in the observational record—comparable to that in the Antarctic ozone hole. Unusually long-lasting cold conditions in the Arctic lower stratosphere led to persistent enhancement in ozone-destroying forms of chlorine and to unprecedented ozone loss, which exceeded 80 per cent over 18–20 kilometres altitude. Our results show that Arctic ozone holes are possible even with temperatures much milder than those in the Antarctic. We cannot at present predict when such severe Arctic ozone depletion may be matched or exceeded.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
References
Farman, J. C., Gardiner, B. G. & Shanklin, J. D. Large losses of total ozone in Antarctica reveal seasonal ClOx/NOx interaction. Nature 315, 207–210 (1985)
Solomon, S., Garcia, R. R., Rowland, F. S. & Wuebbles, D. J. On the depletion of Antarctic ozone. Nature 321, 755–758 (1986)
Molina, L. T. & Molina, M. J. Production of Cl2O2 from the self-reaction of the ClO radical. J. Phys. Chem. 91, 433–436 (1987)
Anderson, J. G., Brune, W. H. & Proffitt, M. H. Ozone destruction by chlorine radicals within the Antarctic vortex: the spatial and temporal evolution of ClO-O3 anticorrelation based on in situ ER-2 data. J. Geophys. Res. 94, 11465–11479 (1989)
Solomon, S. Stratospheric ozone depletion: a review of concepts and history. Rev. Geophys. 37, 275–316 (1999)
Schoeberl, M. R. & Hartmann, D. L. The dynamics of the stratospheric polar vortex and its relation to springtime ozone depletions. Science 251, 46–52 (1991)
World Meteorological Organization . Scientific Assessment of Ozone Depletion: 2010 (Report 52, Global Ozone Research and Monitoring Project, 2011)
Solomon, P. M. et al. High concentrations of chlorine monoxide at low altitudes in the Antarctic spring stratosphere: secular variation. Nature 328, 411–413 (1987)
Waters, J. W. et al. Stratospheric ClO and ozone from the Microwave Limb Sounder on the Upper Atmosphere Research Satellite. Nature 362, 597–602 (1993)
Santee, M. L., Manney, G. L., Waters, J. W. & Livesey, N. J. Variations and climatology of ClO in the polar lower stratosphere from UARS Microwave Limb Sounder measurements. J. Geophys. Res.. 108, 4454, http://dx.doi.org/10.1029/2002JD003335 (2003)
Santee, M. L. et al. A study of stratospheric chlorine partitioning based on new satellite measurements and modeling. J. Geophys. Res.. 113, D12307, http://dx.doi.org/10.1029/2007JD009057 (2008)
World Meteorological Organization . Scientific Assessment of Ozone Depletion: 2006 (Report 50, Global Ozone Research and Monitoring Project, 2007)
Rex, M. et al. Arctic winter 2005: implications for stratospheric ozone loss and climate change. Geophys. Res. Lett.. 33, L23808, http://dx.doi.org/10.1029/2006GL026731 (2006)
Manney, G. L. et al. EOS MLS observations of ozone loss in the 2004–2005 Arctic winter. Geophys. Res. Lett.. 33, L04802, http://dx.doi.org/10.1029/2005GL024494 (2006)
Harris, N. R. P., Lehmann, R., Rex, M. & von der Gathen, P. A closer look at Arctic ozone loss and polar stratospheric clouds. Atmos. Chem. Phys. 10, 8499–8510 (2010)
Rex, M. et al. Arctic ozone loss and climate change. Geophys. Res. Lett.. 31, L04116, http://dx.doi.org/10.1029/2003GL018844 (2004)
Tilmes, S., Müller, R., Engel, A., Rex, M. & Russell, J. M., III Chemical ozone loss in the Arctic and Antarctic stratosphere between 1992 and 2005. Geophys. Res. Lett.. 33, L20812, http://dx.doi.org/10.1029/2006GL026925 (2006)
Poole, L. R. & Pitts, M. C. Polar stratospheric cloud climatology based on Stratospheric Aerosol Measurement II observations from 1978 to 1989. J. Geophys. Res. 99, 13083–13089 (1994)
Fromm, M. D. et al. An analysis of Polar Ozone and Aerosol Measurement (POAM) II Arctic stratospheric cloud observations, 1993–1996. J. Geophys. Res. 104, 24341–24357 (1999)
Pitts, M. C., Poole, L. R. & Thomason, L. W. CALIPSO polar stratospheric cloud observations: second-generation detection algorithm and composition discrimination. Atmos. Chem. Phys. 9, 7577–7589 (2009)
Douglass, A. R. et al. Interhemispheric differences in springtime production of HCl and ClONO2 in the polar vortices. J. Geophys. Res. 100, 13967–13978 (1995)
Manney, G. L. et al. Chemical depletion of ozone in the Arctic lower stratosphere during winter 1992–93. Nature 370, 429–434 (1994)
Tegtmeier, S., Rex, M., Wohltmann, I. & Krüger, K. Relative importance of dynamical and chemical contributions to Arctic wintertime ozone. Geophys. Res. Lett. 35 L17801 http://dx.doi.org/10.1029/2008GL034250 (2008)
Rex, M. et al. In situ measurements of stratospheric ozone depletion rates in the Arctic winter of 1991/1992: a Lagrangian approach. J. Geophys. Res. 103, 5843–5853 (1998)
Manney, G. L. et al. Lagrangian transport calculations using UARS data. Part II: ozone. J. Atmos. Sci. 52, 3069–3081 (1995)
Manney, G. L. et al. Variability of ozone loss during Arctic winter (1991–2000) estimated from UARS Microwave Limb Sounder measurements. J. Geophys. Res.. 108, 4149, http://dx.doi.org/10.1029/2002JD002634 (2003)
Wohltmann, I., Lehmann, R. & Rex, M. The Lagrangian chemistry and transport model ATLAS: simulation and validation of stratospheric chemistry and ozone loss in the winter 1999/2000. Geosci. Model Dev. 3, 585–601 (2010)
von der Gathen, P. et al. Observational evidence for chemical ozone depletion over the Arctic in winter 1991–92. Nature 375, 131–134 (1995)
Gernandt, H. The vertical ozone distribution above the GDR-research base, Antarctica in 1985. Geophys. Res. Lett. 14, 84–86 (1987)
Rex, M. et al. Prolonged stratospheric ozone loss in the 1995–96 Arctic winter. Nature 389, 835–838 (1997)
Manney, G. L., Froidevaux, L., Santee, M. L., Zurek, R. W. & Waters, J. W. MLS observations of Arctic ozone loss in 1996–97. Geophys. Res. Lett. 24, 2697–2700 (1997)
Manney, G. L., Santee, M. L., Froidevaux, L., Waters, J. W. & Zurek, R. W. Polar vortex conditions during the 1995–96 Arctic winter: meteorology and MLS ozone. Geophys. Res. Lett. 23, 3203–3206 (1996)
Petzoldt, K. The role of dynamics in total ozone deviations from their long-term mean over the Northern Hemisphere. Ann. Geophys. 17, 231–241 (1999)
Hood, L. L., Soukharev, B. E., Fromm, M. & McCormack, J. P. Origin of extreme ozone minima at middle to high northern latitudes. J. Geophys. Res. 106, 20925–20940 (2001)
Manney, G. L. et al. Aura Microwave Limb Sounder observations of dynamics and transport during the record-breaking 2009 Arctic stratospheric major warming. Geophys. Res. Lett.. 36, L12815, http://dx.doi.org/10.1029/2009GL038586 (2009)
Newman, P. A. et al. What would have happened to the ozone layer if chlorofluorocarbons (CFCs) had not been regulated? Atmos. Chem. Phys. 9, 2113–2128 (2009)
Newman, P. A., Nash, E. R. & Rosenfield, J. E. What controls the temperatures of the Arctic stratosphere during the spring? J. Geophys. Res. 106, 19999–20010 (2001)
Polvani, L. M. & Saravanan, R. The three-dimensional structure of breaking Rossby waves in the polar wintertime stratosphere. J. Atmos. Sci. 57, 3663–3685 (2000)
Orsolini, Y. J., Karpechko, A. Y. & Nikulin, G. Variability of the Northern Hemisphere polar stratospheric cloud potential: the role of North Pacific disturbances. Q. J. R. Meteorol. Soc. 135, 1020–1029 (2009)
Woollings, T., Charlton-Perez, A., Ineson, S., Marshall, G. & Masato, G. Associations between stratospheric variability and tropospheric blocking. J. Geophys. Res.. 115, D06108, http://dx.doi.org/10.1029/2009JD012742 (2010)
Butchart, N. et al. Multimodel climate and variability of the stratosphere. J. Geophys. Res.. 116, D05102, http://dx.doi.org/10.1029/2010JD014995 (2011)
Charlton-Perez, A. et al. The potential to narrow uncertainty in projections of stratospheric ozone over the 21st century. Atmos. Chem. Phys. 10, 9473–9486 (2010)
Reinecker, M. M. et al. MERRA — NASA’s modern-era retrospective analysis for research and applications. J. Clim. 24 3624–3648 http://dx.doi.org/10.1175/JCLI-D-11-00015.1 (2011)
Hunt, W. H. et al. CALIPSO lidar description and performance assessment. J. Atmos. Ocean. Technol. 26, 1214–1228 (2009)
Waters, J. W. et al. The Earth Observing System Microwave Limb Sounder (EOS MLS) on the Aura satellite. IEEE Trans. Geosci. Rem. Sens. 44, 1075–1092 (2006)
Levelt, P. F. et al. The Ozone Monitoring Instrument. IEEE Trans. Geosci. Rem. Sens. 44, 1093–1101 (2006)
Rex, M. et al. Chemical ozone loss in the Arctic winter 1994/95 as determined by the Match technique. J. Atmos. Chem. 32, 35–59 (1999)
Rex, M. et al. Chemical depletion of Arctic ozone in winter 1999/2000. J. Geophys. Res.. 107, 8276, http://dx.doi.org/10.1029/2001JD000533 (2002)
Hoskins, B. J., McIntyre, M. E. & Robertson, A. W. On the use and significance of isentropic potential-vorticity maps. Q. J. R. Meteorol. Soc. 111, 877–946 (1985)
McPeters, R. D. et al. Earth Probe Total Ozone Mapping Spectrometer (TOMS) Data Products User’s Guide (NASA Technical Publication 1998-206895, 1998)
Manney, G. L., Zurek, R. W., Gelman, M. E., Miller, A. J. & Nagatani, R. The anomalous Arctic lower stratospheric polar vortex of 1992–1993. Geophys. Res. Lett. 21, 2405–2408 (1994)
Manney, G. L. et al. Solar occultation satellite data and derived meteorological products: Sampling issues and comparisons with Aura MLS. J. Geophys. Res.. 112, D24S50, http://dx.doi.org/10.1029/2007JD008709 (2007)
Butchart, N. & Remsberg, E. E. The area of the stratospheric polar vortex as a diagnostic for tracer transport on an isentropic surface. J. Atmos. Sci. 43, 1319–1339 (1986)
Hanson, D. & Mauersberger, K. Laboratory studies of the nitric acid trihydrate: implications for the south polar stratosphere. Geophys. Res. Lett. 15, 855–858 (1988)
Hostetler, C. A. et al. CALIOP algorithm theoretical basis document. Calibration and Level 1 data products (Technical Report, NASA Langley Research Center, 2006); available at 〈 http://www-calipso.larc.nasa.gov/resources/pdfs/PC-SCI-201v1.0.pdf〉.
Livesey, N. J. et al. Version 3.3 Level 2 data quality and description document. (Technical Report JPL D-33509, Jet Propulsion Laboratory, 2010); available at 〈http://mls.jpl.nasa.gov/data/v3-3_data_quality_document.pdf〉.
Smit, H. G. et al. Assessment of the performance of ECC-ozonesondes under quasi-flight conditions in the environmental simulation chamber: insights from the Jülich Ozone Sonde Intercomparison Experiment (JOSIE). J. Geophys. Res.. 112, D19306, http://dx.doi.org/10.1029/2006JD007308 (2007)
Krämer, M. et al. Intercomparison of stratospheric chemistry models under polar vortex conditions. J. Atmos. Chem. 45, 51–77 (2003)
McPeters, R. et al. Validation of the Aura Ozone Monitoring Instrument total column ozone product. J. Geophys. Res.. 113, D15S14, http://dx.doi.org/10.1029/2007JD008802 (2008)
Acknowledgements
We thank the MLS (especially A. Lambert, D. Miller, W. Read, M. Schwartz, P. Stek, J. Waters), OMI (especially P. K. Bhartia, G. Jaross, G. Labow), CALIPSO and Match science teams, as well as A. Douglass, J. Joiner and the Aura project, for their support. We also thank W. Daffer and R. Fuller for programming assistance at JPL; the many observers whose work went into obtaining the ozone-sonde measurements; the ozone scientists who participated in the discussion of the 2011 Arctic ozone loss and appropriate definition of an Arctic ozone hole (including, but not limited to, N. Harris, G. Bodeker, G. Braathen, M. Kurylo, R. Salawitch); and especially P. Newman and K. Minschwaner for discussions and comments. Meteorological analyses were provided by NASA’s Global Modeling and Assimilation Office (GMAO) and by the European Centre for Medium-Range Weather Forecasts. We thank S. Pawson of GMAO for advice on usage of the MERRA reanalysis. Ozone-sonde measurements at Alert, Eureka, Resolute Bay, Churchill and Goose Bay were funded by Environment Canada. Additional ozone sondes were flown at Eureka as part of the Canadian Arctic Atmospheric Chemistry Experiment (ACE) Validation Campaign and were funded by the Canadian Space Agency. Academy of Finland provided partial funding for performing and processing ozone-sonde measurements in Jokioinen and Sodankylä. Ozone soundings and work at AWI were partially funded by the EC DG Research through the RECONCILE project. Work at the Jet Propulsion Laboratory, California Institute of Technology, and at Science Systems and Applications Inc., was done under contract with NASA.
Author information
Authors and Affiliations
Contributions
G.L.M. and M.L.S. led analysis of MLS data; M.R. led analysis of ozone-sonde data; G.L.M. led the meteorological data analysis. M.R., G.L.M., N.J.L. and I.W. did chemical ozone loss calculations. R.L. and M.R. performed and analysed chemical box model calculations. M.C.P. and L.R.P. provided CALIPSO/CALIOP data analyses; E.R.N. and P.V. provided TOMS and OMI data analyses. L.F., M.L.S., G.L.M. and N.J.L. provided expertise on MLS data usage; D.P.H., P.V. and P.F.L. provided expertise on OMI data usage. J.D., V.D., H.G., B.J., R.K., E.K., N.L., A.M., C.T.M., H.N., M.C.P., D.W.T., P.v.d.G., K.A.W. and N.S.Z. were responsible for performing and processing ozone-sonde measurements. All authors contributed comments on the manuscript. G.L.M., M.L.S. and M.R. jointly compiled and synthesized the results. G.L.M. and M.L.S. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
CALIOP data are publicly available at http://eosweb.larc.nasa.gov/PRODOCS/calipso/table_calipso.html, MLS data at http://disc.sci.gsfc.nasa.gov/Aura/data-holdings/MLS, OMI data at http://disc.sci.gsfc.nasa.gov/Aura/data-holdings/OMI/omto3_v003.shtml, and GEOS-5 MERRA analyses through http://disc.sci.gsfc.nasa.gov/mdisc/data-holdings/merra/. The balloon-borne Antarctic ozone-sonde data recorded in 1985 and the following years are publicly available at http://dx.doi.org/10.1594/PANGAEA.547983.
Supplementary information
Supplementary Information
The file contains a Supplementary Discussion, Supplementary Figures 1-7 with legends and additional references. (PDF 2872 kb)
Supplementary Data 1A
The data shows Arctic Ozone Data (ZIP 5905 kb)
Supplementary Data 1B
The data shows Arctic Ozone Station Information (XLS 10 kb)
Rights and permissions
About this article
Cite this article
Manney, G., Santee, M., Rex, M. et al. Unprecedented Arctic ozone loss in 2011. Nature 478, 469–475 (2011). https://doi.org/10.1038/nature10556
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10556
This article is cited by
-
The impact of boreal spring stratospheric final warming on surface air temperature over Northern Hemisphere in ERA5 and CMIP6 models
Climate Dynamics (2024)
-
No evidence of worsening Arctic springtime ozone losses over the 21st century
Nature Communications (2023)
-
Dependence of column ozone on future ODSs and GHGs in the variability of 500-ensemble members
Scientific Reports (2023)
-
Reply to: No evidence of worsening Arctic springtime ozone losses over the 21st century
Nature Communications (2023)
-
The Influence of Meridional Variation in North Pacific Sea Surface Temperature Anomalies on the Arctic Stratospheric Polar Vortex
Advances in Atmospheric Sciences (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
