
Abstract
Using DNA extracted from a finger bone found in Denisova Cave in southern Siberia, we have sequenced the genome of an archaic hominin to about 1.9-fold coverage. This individual is from a group that shares a common origin with Neanderthals. This population was not involved in the putative gene flow from Neanderthals into Eurasians; however, the data suggest that it contributed 4–6% of its genetic material to the genomes of present-day Melanesians. We designate this hominin population ‘Denisovans’ and suggest that it may have been widespread in Asia during the Late Pleistocene epoch. A tooth found in Denisova Cave carries a mitochondrial genome highly similar to that of the finger bone. This tooth shares no derived morphological features with Neanderthals or modern humans, further indicating that Denisovans have an evolutionary history distinct from Neanderthals and modern humans.
Similar content being viewed by others



Main
Less than 200,000 years ago, anatomically modern humans (that is, humans with skeletons similar to those of present-day humans) appeared in Africa. At that time, as well as later when modern humans appeared in Eurasia, other ‘archaic’ hominins were already present in Eurasia. In Europe and western Asia, hominins defined as Neanderthals on the basis of their skeletal morphology lived from at least 230,000 years ago before disappearing from the fossil record about 30,000 years ago1. In eastern Asia, no consensus exists about which groups were present. For example, in China, some have emphasized morphological affinities between Neanderthals and the specimen of Maba2, or between Homo heidelbergensis and the Dali skull3. However, others classify these specimens as ‘early Homo sapiens’4. In addition, until at least 17,000 years ago, Homo floresiensis, a short-statured hominin that seems to represent an early divergence from the lineage leading to present-day humans5,6,7, was present on the island of Flores in Indonesia and possibly elsewhere.
DNA sequences retrieved from hominin remains offer an approach complementary to morphology for understanding hominin relationships. For Neanderthals, the nuclear genome was recently determined to about 1.3-fold coverage8. This revealed that Neanderthal DNA sequences and those of present-day humans share common ancestors on average about 800,000 years ago and that the population split of Neanderthal and modern human ancestors occurred 270,000–440,000 years ago. It also showed that Neanderthals shared more genetic variants with present-day humans in Eurasia than with present-day humans in sub-Saharan Africa, indicating that gene flow from Neanderthals into the ancestors of non-Africans occurred to an extent that 1–4% of the genomes of people outside Africa are derived from Neanderthals8. In addition, ten partial and six complete mitochondrial (mt)DNA sequences have been determined from Neanderthals9,10,11,12,13,14,15,16,17. This has shown that all Neanderthals studied so far share a common mtDNA ancestor on the order of 100,000 years ago10, and in turn, share a common ancestor with the mtDNAs of present-day humans about 500,000 years ago10,18,19 (as expected, this is older than the Neanderthal–modern human population split time of 270,000–440,000 years ago estimated from the nuclear genome8). One of these mtDNA sequences has also shown that hominins carrying mtDNAs typical of Neanderthals were present as far east as the Altai Mountains in southern Siberia13.
In 2008, the distal manual phalanx of a juvenile hominin was excavated at Denisova Cave. This site is located in the Altai Mountains in southern Siberia, and is a reference site for the Middle to Upper Palaeolithic of the region where systematic excavations over the past 25 years have uncovered cultural layers indicating that human occupation at the site started up to 280,000 years ago20. The phalanx was found in layer 11, which has been dated to 50,000 to 30,000 years ago. This layer contains microblades and body ornaments of polished stone typical of the ‘Upper Palaeolithic industry’ generally thought to be associated with modern humans, but also stone tools that are more characteristic of the earlier Middle Palaeolithic, such as sidescrapers and Levallois blanks21,22,23.
Recently, we used a DNA capture approach10 in combination with high-throughput sequencing to determine a complete mtDNA genome from the Denisova phalanx. Surprisingly, this mtDNA diverged from the common lineage leading to modern human and Neanderthal mtDNAs about one million years ago19, that is, about twice as far back in time as the divergence between Neanderthal and modern human mtDNAs. However, mtDNA is maternally inherited as a single unit without recombination, and therefore is subject to chance events such as genetic drift, as well as gene flow and positive selection. In contrast, the nuclear genome comprises tens of thousands of unlinked, mostly neutrally evolving loci. This allows for analyses of genetic relationships that are robust to the stochasticity of genetic drift, and are much less affected by positive selection. To clarify the relationship of the Denisova individual to other hominin groups, we have therefore sequenced the Denisova nuclear genome and analysed its genomic relationships to Neanderthals and present-day humans. We have also attempted to clarify the chronology of hominin occupation of the cave and have identified a tooth from this group of hominins among material excavated in Denisova Cave.
DNA sequence determination
The entire internal portion of the phalanx sample was used for DNA extraction in our clean-room facility, where procedures to minimize contamination from present-day human DNA are rigorously implemented24,25 (Supplementary Information section 1). The DNA was treated with two enzymes: uracil-DNA-glycosylase, which removes uracil residues from DNA to leave abasic sites26, and endonuclease VIII, which cuts DNA at the 5′ and 3′ sides of abasic sites. Subsequent incubation with T4 polynucleotide kinase and T4 DNA polymerase was used to generate 5′-phosphorylated blunt ends that are amenable to adaptor ligation. Because the great majority of uracil residues occur close to the ends of ancient DNA molecules, this procedure leads to only a moderate reduction in average length of the molecules in the library, but a several-fold reduction in uracil-derived nucleotide misincorporation27.
Two independent sequencing libraries (SL3003 and SL3004) were created from the DNA, using a modified Illumina protocol28 where a polymerase chain reaction (PCR) is used to add a 7-nucleotide index (in this case 5′-GTCGACT-3′) to the library molecules. This index ensures that the libraries are not contaminated by other sequencing libraries when they are taken out of the clean room to be sequenced29. The libraries were sequenced on the Illumina Genome Analyser IIx platform for 101 cycles from each end of the molecules and an additional 7 cycles for determination of the index until almost every unique sequence in the libraries had been seen multiple times, that is, almost every clone present in the libraries has been sequenced (Supplementary Information section 1). Bases were called using the machine-learning algorithm Ibis30 and an overlap of at least 11 bases was required for paired-end reads to be fused to full-molecule-size DNA sequences that were further analysed. This results in a greatly reduced error rate27, although it removes the few molecules that are above 191 nucleotides in length from analysis (∼0.1% in SL3003 and ∼0.2% in SL3004). Sequences were mapped using the program BWA31 to the human (hg18/NCBI 36) and the chimpanzee (panTro2/CGSC 2.1) genomes as well as to the inferred ancestral genome of these species (from the six-way Enredo-Pecan-Ortheus alignment)32. PCR duplicates were identified and used to further increase sequence accuracy by calling consensus sequences.
A total of 82,227,320 sequences mapped uniquely (mapping quality ≥30) to the human genome, yielding about 5.2 gigabases of DNA sequences (1.9-fold genomic coverage), and 72,304,848 sequences mapped uniquely to the chimpanzee genome. When the substitutions inferred to have occurred on the Denisova and the present-day human lineages were compared, the relative numbers of different classes of nucleotide substitutions are remarkably similar, and the excess number of candidate substitutions on the Denisova lineage relative to the present-day human lineage is only 1.7-fold (Supplementary Fig. 2.2 and Supplementary Table 2.4). This reflects an improvement in error rate over the Neanderthal genome by over an order of magnitude8 and is mainly due to the enzymatic removal of uracil residues from the Denisova DNA27. We estimate that most errors in the Denisova DNA sequences are due to low genomic coverage rather than to any features typical of ancient DNA.
Human DNA contamination estimates
Although rigorous measures to prevent contamination of the experiments by DNA from present-day humans were implemented at all laboratory steps, it is impossible to completely prevent contamination because bone samples as well as reagents may be contaminated before they enter the clean-room facility. To estimate the levels of contamination in the sequences produced we used three approaches (Supplementary Information section 3).
First, we estimated the level of mtDNA contamination using 276 sequence positions where the Denisova mtDNA differs from >99% of present-day human mtDNAs. For library SL3003, we observed 7,433 unique sequences that covered such positions and 7,421 were of the Denisova type. For library SL3004 the corresponding numbers were 5,042 and 5,036, indicating that the mtDNA contamination in the libraries is on the order of 0.2% (95% confidence interval (CI): 0.1–0.3%) and 0.1% (CI: 0.1–0.3%), respectively.
Second, we identified sequences that are unique to the Y chromosome8. If the individual from whom the phalanx derives is female, the number of such sequences represents the extent of male DNA contamination. We found zero and three such Y chromosomal sequences in the two libraries, respectively, whereas 1,449 and 696 are expected if the individual is male. Thus, the bone derives from a female and male DNA contamination in the two libraries is on the order of 0.00% (CI: 0.00–0.25%) and 0.43% (CI: 0.09–1.26%), respectively.
Third, to estimate the extent of nuclear DNA contamination we used one library to identify positions where the Denisova individual carries an ancestral, that is, chimpanzee-like, sequence variant that among present-day humans is derived and not known to vary. We then examined sequences that map at these positions in the other library and determined if they carry the ancestral sequence or the derived sequence. Observation of a derived sequence in the second library could be due to one of three possibilities: that the DNA fragment in question comes from present-day human contamination; that the Denisova individual is heterozygous at the position in question; or that there has been a sequencing error. We implemented a maximum likelihood method that uses the number of independent observations of ancestral and derived states across positions to co-estimate contamination along with heterozygosity and sequencing error as nuisance parameters (Supplementary Information section 3). From this analysis, both libraries are inferred to have contamination rates of less than 1%.
Ancestral features and duplications
The Denisova draft genome sequence allows features that are ancestral in the Denisova genome and derived in present-day humans to be identified. We previously described a set of 10.5 million single nucleotide differences and about half a million insertion/deletions (indels) inferred to be due to changes that occurred on the human lineage since the split from the common ancestor with the chimpanzee8. Of these, 4,267,431 (40.5%) single nucleotide differences and 105,372 (22.0%) indels are covered by the Denisova sequences. We identified 129 inferred amino substitutions and 14 indels in the coding sequences of genes where the Denisova individual carries the ancestral alleles at positions where present-day humans carry derived alleles and are not known to vary (Supplementary Information section 4). We also identified 90 such sites in 5′ untranslated regions (UTRs), 392 in 3′ UTRs, two in microRNA genes and 104 in human accelerated regions. When we compared the Denisova and Neanderthal genomes we found that they carry the same assigned state at single nucleotide differences in 87.9% of the ancestral positions and 97.7% of the derived positions. The results for indels are similar: 87.6% for ancestral states and 98.6% for the derived states (Supplementary Table 4.3).
We analysed the segmental duplication content of the Denisova genome by detecting regions with an excess read depth (Supplementary Information section 5). In a three-way comparison of Denisova, Neanderthal and present-day human genomes, we found an excess of private Denisova duplications (2.27 megabases (Mb)) compared with duplications that were private in Neanderthals (0.60 Mb) or present-day humans (1.32 Mb). These regions were identified based on signatures of both excess read depth and increased sequence divergence, making them unlikely to be artefacts. We also identified two regions where the duplication architecture of Denisova is more similar to that of chimpanzee than to that of either Neanderthals or present-day humans, including two chromosomal regions associated with neurological disease in humans: spinal muscular atrophy on 5q13 (including SMN2, one of the most recent gene duplications in the human lineage) and neuropsychiatric disease on 16p12.1.
Relationship to Neanderthals and modern humans
A fundamental question is whether the Denisova individual is an outgroup to Neanderthals and modern humans, as the mtDNA suggests19, whether it is a sister group to Neanderthals or to modern humans, or whether it falls within the range of variation of either of these two groups. We addressed this by estimating the divergence between the Denisova and the human genome reference sequence as a fraction of the divergence between present-day humans and the common ancestor shared with the chimpanzee. To do this, we scored the frequency with which the Denisova genome carries the human versus the chimpanzee state at positions where the human and chimpanzee reference genomes differ; assuming constant evolutionary rates (Supplementary Information section 2). We restricted this analysis to the parts of the human reference genome that are of African ancestry33 as gene flow from Neanderthals to non-Africans8 could otherwise complicate these analyses. The Denisova genome diverged from the reference human genome 11.7% (CI: 11.4–12.0%) of the way back along the lineage to the human–chimpanzee ancestor. For the Vindija Neanderthal, the divergence is 12.2% (CI: 11.9–12.5%). Thus, whereas the divergence of the Denisova mtDNA to present-day human mtDNAs is about twice as deep as that of Neanderthal mtDNA19, the average divergence of the Denisova nuclear genome from present-day humans is similar to that of Neanderthals.
A possible explanation for the similar divergence of the Denisova individual and Neanderthals from present-day Africans is that they both descend from a common ancestral population that separated earlier from ancestors of present-day humans. Such a scenario would predict a closer relationship between the Denisova individual and Neanderthals than between either of them and present-day humans. To test this prediction, we estimated the divergence between pairs of seven ancient and modern genomes (Denisova, Neanderthals, French, Han, Papuan, Yoruba and San), using an approach where we correct for error rates in each genome based on the assumption that each has the same number of true differences from chimpanzee (Supplementary Information section 6). The average divergence between Denisova and Vindija Neanderthals is estimated to be 9.84% of the way to the chimpanzee–human ancestor; that is, less than the average 12.38% divergence of both from present-day Africans. Assuming 6.5 million years for human–chimpanzee divergence, this implies that DNA sequences of Neanderthals and the Denisova individual diverged on average 640,000 years ago, and from present-day Africans 804,000 years ago.
To analyse further the relationship of the Denisova individual and Neanderthals, we aligned Denisova, Neanderthal and Yoruba sequences to the chimpanzee genome, picked a single sequence at random to represent each group, and examined sites where two copies of a derived and one copy of an ancestral allele were observed. Sequencing errors are expected to make a negligible contribution at such sites. The number of sites where the Denisova individual and Neanderthal cluster to the exclusion of the Yoruba and chimpanzee is 46,362, compared with an average of 22,012 sites for the other two possible patterns (Yoruba and Denisova, or Yoruba and Neanderthal). This excess of sites where Denisova and Neanderthal cluster supports the view that the Denisova individual and Neanderthals share a common history since separating from the ancestors of modern humans (Supplementary Information section 6).
A Neanderthal-specific bottleneck
The fact that the Denisova nuclear genome on average shares a more recent common ancestor with Neanderthal than with present-day humans raises the question of whether the overall DNA sequence divergence of the Denisova individual falls inside the group morphologically and geographically defined as Neanderthals, or if it represents a sister group to Neanderthals.
To investigate this question, we took advantage of the fact that in addition to the three individuals from Vindija Cave, Croatia, from which most of the Neanderthal genome sequences were produced, we have determined nuclear DNA sequences from three further Neanderthal individuals from Russia, Spain and Germany8. Of these, the 60,000–70,000-year-old skeleton of a Neanderthal child found in Mezmaiskaya Cave, Russia, is both oldest and geographically closest to the Denisova individual. Using the 56 Mb of autosomal DNA sequences determined from this specimen8, we estimate that the DNA sequence divergence between the Vindija and Mezmaiskaya Neanderthals corresponds to a date of 140,000 ± 33,000 years ago (Supplementary Information section 6) (Fig. 1). This remarkably low divergence—which is about one-third of the closest pair of present-day humans that we analysed—is in agreement with the observation that diversity among Neanderthal mtDNAs is low relative to present-day humans10 and indicates that the Vindija and Mezmaiskaya Neanderthals descend from a common ancestral population that experienced a drastic bottleneck since separating from the ancestors of the Denisova individual.

Vindija 33.16, Vindija 33.25 and Vindija 33.26 refer to the catalogue numbers of the Neanderthal bones.
To understand further the bottleneck in the history of Vindija and Mezmaiskaya Neanderthals, we examined four-way alignments of the Vindija Neanderthal genome sequence, the Mezmaiskaya Neanderthal, the Denisova individual and the chimpanzee genome. At transversion substitutions where two copies of the derived alleles are observed, we detect 924 substitutions that cluster the Vindija and Mezmaiskaya Neanderthals, 80 that cluster Vindija and Denisova, and 81 that cluster Mezmaiskaya and Denisova. This corresponds to at least a 65% probability that the DNA sequences in the Neanderthals share a common ancestor more recently than their split from the ancestor of the Denisova individual (Supplementary Information section 7). It is much higher than the 15–20% probability associated with the ‘Out of Africa’ bottleneck common to present-day non-Africans34. If we replace the Mezmaiskaya Neanderthal in this analysis with a Neanderthal from El Sidron, Spain, or from Feldhofer, Germany, results are qualitatively similar although numbers are smaller (Supplementary Information section 7). Thus, we conclude that late Neanderthals across a broad geographical range have a population history distinct from that of the Denisova individual in that they share a strong population bottleneck not experienced by the ancestors of the Denisova individual. We call the group to which this individual belonged Denisovans in analogy to Neanderthals, as Denisovans are described for the first time based on molecular data from Denisova Cave just as Neanderthals were first described based on skeletal remains retrieved in the Neander Valley in Germany.
No Denisovan gene flow into all Eurasians
We have previously shown that Vindija Neanderthals share more derived alleles with non-Africans than with Africans, consistent with Neanderthals contributing 1–4% of the genomes of present-day humans across Eurasia8. To investigate the extent to which the Denisova individual shares this pattern, we examined alignments of sets of four genomes, each consisting of an African (Yoruba or San), a Eurasian (French or Han), an archaic hominin (Neanderthal or Denisovan) and the chimpanzee. We randomly sampled one allele from each of the three hominins, and counted all transversion differences between the African and the Eurasian where the archaic individual carries the derived allele (the ‘D statistics’ of ref. 8). Neanderthals match the French genome on average 4.6 ± 0.7% more often than they match the Yoruba genome (Table 1). Although the Denisova individual also matches the French more than the Yoruba genome, this skew is significantly less strong at 1.8 ± 0.5%. The estimates of D statistics were quantitatively consistent (within two standard deviations) for all other choices of Eurasian and African populations (Table 1). These findings indicate that the archaic component of the Eurasian gene pool is less closely related to the Denisova individual than to Neanderthals.
We also examined 13 genomic regions that we previously identified as candidates for a contribution of archaic genetic material into non-Africans, based on their deeper genetic divergences in non-Africans than in Africans8. Using ‘tag SNPs’ that are informative about whether a haplotype is from the lineage unique to non-Africans, we find that the Denisova individual matches the deeply diverged non-African haplotype in 6 cases, whereas Neanderthals do so in 11 cases (Supplementary Information section 7). Thus, both Neanderthals and Denisovans are more related than would be expected by chance to these genomic segments, but the signal in Denisovans is weaker.
These analyses indicate that Neanderthals are more closely related than Denisovans to the population that contributed to the gene pool of the ancestors of present-day Eurasians. The fact that Eurasians share some additional affinity with the Denisova individual relative to Africans is compatible with a scenario in which Denisovans shared some of their history with Neanderthals before the gene flow from Neanderthals into modern humans occurred.
Denisovan gene flow into the ancestors of Melanesians
Although the Denisova individual derives from a population that was not directly involved in the gene flow from Neanderthals to Eurasians, it is possible that Denisovans admixed with the ancestors of present-day people in some parts of the Old World. To investigate this, we analysed the relationship of the Denisova genome to the genomes of 938 present-day humans from 53 populations that have been genotyped at 642,690 single nucleotide polymorphisms (SNPs)35. We scored each of these present-day humans based on their relative proximity to Neanderthals and the Denisova individual at positions where we have high-quality data for both the Neanderthal and Denisova genomes (Supplementary Information section 8). Using the means of the 53 populations, the first two principal components separate the populations into three groups (Fig. 2): first, the 7 sub-Saharan African populations; second, a group of 44 non-African populations as well as one north African group; and third, Papuan and Bougainville populations from Melanesia. When individuals from selected populations are analysed separately, the Papuan and Bougainville islanders remain distinct from almost all individuals outside Africa (Supplementary Fig. 8.1b). Thus, with respect to their relationship to Neanderthals and Denisovans, the Melanesian populations stand out relative to other non-African populations.
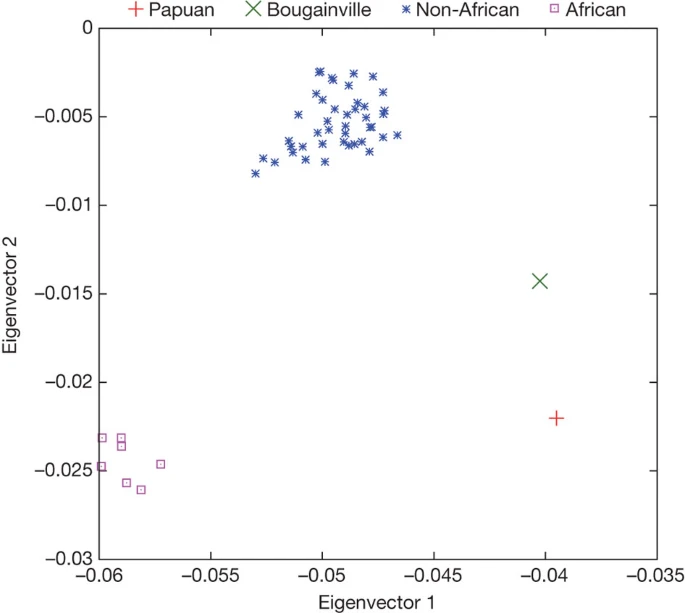
Principal component analysis of the means of 53 present-day human populations projected onto the top two principal components defined by Denisova, Neanderthal and chimpanzee. The seven ‘African’ populations are San, Mbuti, Biaka, Bantu Kenya, Bantu South Africa, Yoruba and Mandenka; the ‘Non-African’ populations are 44 diverse groups from outside Africa except for Papuan and Bougainville islanders.
To explore this further, we analysed the relationship of the Denisova genome to the genomes of five present-day humans that we previously sequenced to about fivefold coverage8 (a Yoruba and a San genome from Africa, a French genome from Europe, a Han genome from China and a Papuan genome from Melanesia), as well as seven present-day humans that we sequenced to 1–2-fold coverage for this study (a Mbuti genome from Africa, a Sardinian genome from Europe, a Mongolian genome from Central Asia, a Cambodian genome from South-East Asia, an additional Papuan genome from Melanesia, a Bougainville islander genome from Melanesia, and a Karitiana genome from South America) (Supplementary Information section 9). We used the D statistic8 to test if various pairs of present-day humans share equal numbers of derived alleles with the Denisova individual. To do this, we restricted comparisons to pairs of present-day humans sequenced at the same time to minimize the chance that differences in sample processing could affect the results. We find that the fivefold coverage Papuan individual shares 4.0 ± 0.7% more alleles with the Denisova individual than does the French individual, and we observed a similar skew in all 10 comparisons of Melanesian and other non-African populations (Table 1). When we stratified the data by base substitution class and chromosome, the D statistics are qualitatively unchanged (Supplementary Information section 10). Similarly, the D statistics are consistent for all depths of read coverage, indicating that mapping errors, for example due to segmental duplications, are not likely to explain these results. Finally, differences in sequencing error rate across samples cannot explain the observed D statistics (Supplementary Information section 10).
Under the assumption that gene flow explains these observations, we determined the direction of this gene flow by asking whether Melanesians and other Eurasians share derived alleles with Africans equally often. If the gene flow was entirely into the ancestors of the Denisovan individual, we would not expect this to affect the relationship of Africans to Melanesians and other Eurasians and thus we would expect them to share derived alleles equally often with Africans. However, we find that derived alleles in Africans match Melanesians 3.4 ± 0.4% less often than other non-Africans (Z = 10.8). Because this skew is seen without using Denisovan data it cannot be explained by gene flow into Denisovans or, for example, by contamination of the Denisova sample by present-day Melanesian DNA. Thus, at least some of the putative gene flow must have been into Melanesians (Supplementary Information section 8).
When we compare the skew in the fraction of derived alleles shared with the two archaic hominins to what would be expected for individuals of 100% Neanderthal or Denisova ancestry, respectively (Supplementary Information section 8 and ref. 8), we estimate that 2.5 ± 0.6% of the genomes of non-African populations derive from Neanderthals, in agreement with our previous estimate of 1–4%8. In addition, we estimate that 4.8 ± 0.5% of the genomes of Melanesians derive from Denisovans. Altogether, as much as 7.4 ± 0.8% of the genomes of Melanesians may thus derive from recent admixture with archaic hominins.
A model of population history
To understand the implications of the relationships observed among the Denisova individual, the Neanderthals and present-day humans, we fit the D statistics described in the previous sections to a parameterized model of population history. The D statistics for the Denisova individual differ in two important ways from those for the Neanderthal. First, the Denisova individual shares fewer derived alleles with either the French or Han Chinese populations than do the Neanderthals. Second, the Denisova individual shares more derived alleles with the Papuans than do the Neanderthals. We are able to fit the data with a model that assumes the Denisovans are a sister group of Neanderthals with a population divergence time of one-half to two-thirds of the time to the common ancestor of Neanderthals and humans. After the divergence of the Denisovans from Neanderthals, there was gene flow from Neanderthals into the ancestors of all present-day non-Africans. Later there was admixture between the Denisovans and the ancestors of Melanesians that did not affect other non-African populations. This model is illustrated in Fig. 3 and is described in detail in Supplementary Information section 11.
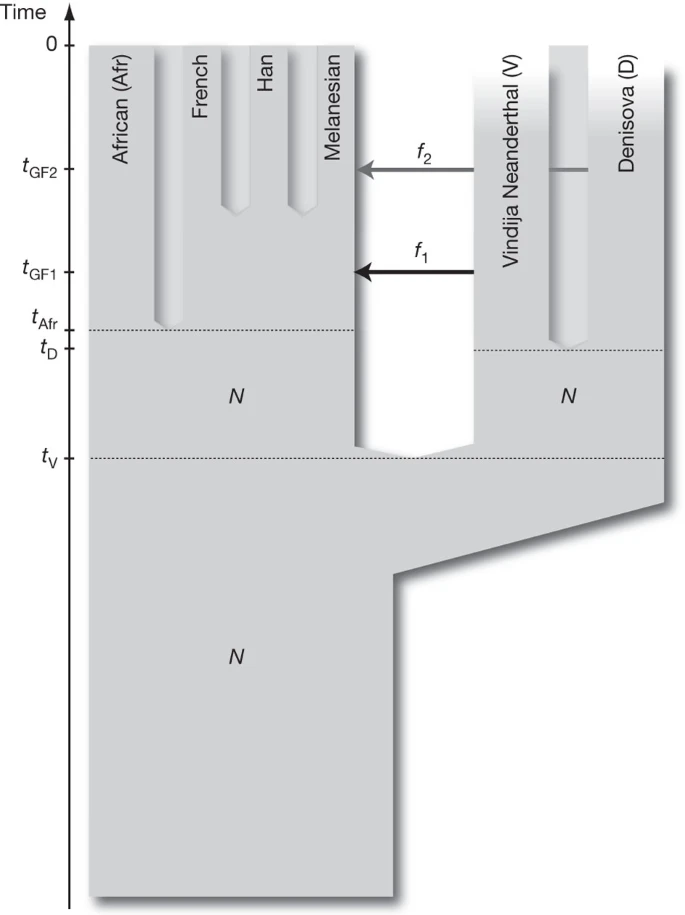
N denotes effective population size, t denotes time of population separation, f denotes amount of gene flow and tGF denotes time of gene flow.
Other, more complex models could also explain the data. For example, a model that invokes only gene flow from Denisovans to Melanesian ancestors outside Africa and assumes four subpopulations in Africa that existed between the times of the origin of Denisovan and Neanderthal ancestors and the ancestors of present-day Eurasians could also fit the data (Supplementary Fig. 11.4). However, because barriers to gene flow between such subpopulations would have to persist for hundreds of thousands of years to create the observed patterns, such a model is less plausible on biological grounds than a model that invokes two instances of gene flow outside Africa.
Discordance of mtDNA and nuclear histories
The population history indicated by the nuclear genome is different from that indicated by the mtDNA phylogeny. There are two possible explanations for this. One is that the mtDNA lineage was introduced into Denisovan ancestors by admixture from another hominin lineage for which we have no data. The other is that the discordance is the result of ‘incomplete lineage sorting’, that is, the random assortment of genetic lineages due to genetic drift which may have allowed a divergent mtDNA lineage to survive in Denisovans by chance while becoming lost in Neanderthals and modern humans. A large ancestral population size makes incomplete lineage sorting more likely to occur. In Supplementary Information section 11, we show that given reasonable assumptions about the size of the ancestral populations, the discordance of the mtDNA phylogeny with that indicated by the nuclear DNA can be explained either by a small amount of admixture from another archaic hominin or by incomplete lineage sorting. Thus, the data do not allow us to favour one hypothesis over the other.
A tooth from Denisova Cave
In 2000, a hominin tooth was discovered in layer 11.1 of the south gallery of Denisova Cave (Fig. 4a, b). The tooth is from a young adult and therefore from another individual than the phalanx which stems from a juvenile (Supplementary Information section 12). To elucidate the relationship of the tooth to the individual from which the phalanx is derived, we extracted DNA from 50 mg of dentin from the root of the tooth and prepared a sequencing library (Supplementary Information section 13). About 0.17% of random DNA sequences determined from this library aligned to the human genome, whereas the rest is likely to represent microbial contamination common in ancient bones. We therefore used a novel DNA capture approach36 to isolate mtDNA sequences from the sequencing library. A total of 15,094 sequences were identified which allowed the complete mtDNA genome to be assembled at an average coverage of 58-fold. This sequence differs at two positions from the mtDNA of the phalanx whereas it differs at about 380 positions from both Neanderthal and present-day humans. The time since the most recent common ancestor of the two mtDNAs from Denisova Cave is estimated to be 7,500 years, with a 95% upper bound of 16,000 years (Supplementary Information section 13). We conclude that the tooth and the phalanx derive from two different individuals that are probably from the same hominin population.
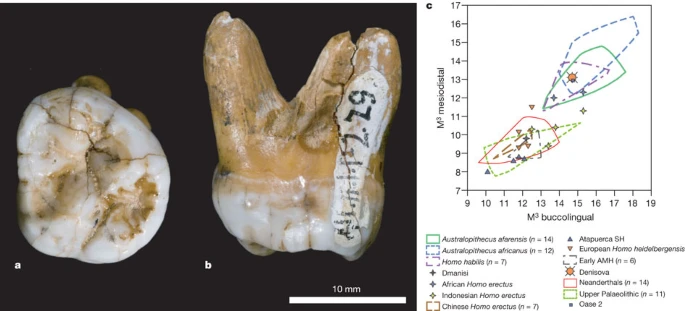
a, b, Occlusal (a) and mesial (b) views. c, Comparison of the Denisova molar to diverse third molars, in a biplot of the mesiodistal and buccolingual lengths (in mm). AMH, anatomically modern humans; SH, Sima de los Huesos. Supplementary Fig. 12.1 presents a similar comparison to second molars.
Morphology of the Denisova molar
The tooth is an almost complete left, probably third, but possibly second, upper molar (Fig. 4b). The crown is trapezoidal and tapers strongly distally, with bulging lingual and buccal walls giving the tooth an inflated appearance (Supplementary Information section 12). The roots are short but robust and strongly flaring.
Overall, the tooth is very large (mesiodistal diameter, 13.1 mm; buccolingual, 14.7 mm). As a third molar, it is outside the range of normal size variation of all fossil taxa of the genus Homo, with the exception of H. habilis and H. rudolfensis, and comparable to Australopithecines (Fig. 4c). Compared to second molars, it is larger than Neanderthals or early modern humans, but similar to H. erectus and H. habilis (Supplementary Fig. 12.1).
Besides size, it is also distinguished from most Neanderthal third molars by the absence of hypocone reduction, and from both second and third Neanderthal molars by the presence of a large talon basin and the strong flare of the crown. Furthermore, it lacks the lingual hypocone projection seen in all Neanderthal first and many second molars, and has strongly diverging roots, unlike the closely spaced and frequently fused roots of Neanderthals.
It is of particular interest to compare the Denisova molar to Middle Pleistocene hominins from China, where H. erectus and other archaic forms, sometimes interpreted as H. heidelbergensis, may have survived until recently. Unfortunately, very few of these fossils preserve third upper molars. Of the few examples that are available, most differ from the Denisova molar by their strongly reduced size. Second molars are more frequent than third molars, and most have a trapezoidal shape like Denisova, but they do not have the lingually skewed position of the hypocone and metacone and the strong basal flare of the crown.
The Denisova molar supports the DNA evidence that the Denisovan population is distinct from late Neanderthals as well as from modern humans. In fact, the primitive traits of the Denisova tooth suggest that Denisovans may have been separated from the Neanderthal lineage before Neanderthal dental features are documented in Western Eurasia (>300,000 years bp) (Supplementary Information section 12), although we cannot exclude the possibility that the Denisovan dental morphology results from a reversion.
Stratigraphy and dating
The small size of both the phalanx and the tooth precludes direct radiocarbon dating. We instead dated seven bone fragments found close to the hominin remains in layer 11 in the east and south galleries. To ensure that they were associated with human occupation of the cave we chose bones that have evidence of human modification, including a rib with regular incisions and a bone projectile point blank generally associated with Upper Palaeolithic cultural assemblages. In the south gallery, where modified bones were not available, we used herbivore bones (Supplementary Information section 12).
Four of the seven dates are infinite dates older than 50,000 years bp (uncalibrated), whereas three are finite dates between 16,000 and 30,000 years bp (Supplementary Table 12.1). The rib with incisions and the projectile point blank are about 30,000 and 23,000 years bp, respectively. Together with three previous dates23 this shows that layer 11 contains cultural remains from at least two different time periods, one period older than 50,000 years bp and one more recent period. However, the stratigraphy is complicated by the discovery of a wedge-shaped area close to the area where the phalanx was found that is likely to be disturbed (Supplementary Information section 12). Hominin remains large enough to allow direct radiocarbon dates may eventually be discovered in the cave, but a reasonable hypothesis is that the phalanx and molar belong to the older occupation.
Discussion
The molecular preservation of the Denisova phalanx is exceptional in that the fraction of endogenous relative to microbial DNA is about 70%. By contrast, in all Neanderthal remains studied so far the relative abundance of endogenous DNA is below 5%, and typically below 1%. Furthermore, the average length of hominin DNA fragments in the Denisova phalanx is 58 base pairs (bp) (SL3003) and 74 bp (SL3004) in spite of the enzymatic treatment that removes uracil residues and decreases the average fragment size, whereas in most well-preserved Neanderthal samples it is 50 bp or smaller without this treatment. Thus, although many Neanderthals are preserved under conditions apparently similar to those in Denisova Cave, the Denisova phalanx is one of few bones found in temperate conditions that are as well preserved as many permafrost remains37,38. It is not clear why this is. It is not due to some condition that affects all hominin remains in Denisova Cave because the fraction of endogenous DNA in the tooth is 0.17%; that is, typical of other Late Pleistocene hominin remains. It is possible that a rapid desiccation of the tissue after death, which would limit degradation of the DNA by endogenous enzymes as well as microbial growth, has allowed this exceptional preservation.
The Denisova individual and the population to which it belonged carry some exceptionally archaic molecular (mtDNA) as well as morphological (dental) features. Nevertheless, the picture that emerges from analysis of the nuclear genome is one where the Denisova population is a sister group to Neanderthals. Three possibilities could account for how such archaic features have come to be present in Denisovans. One possibility is that these features were retained in Denisovans but became lost in modern humans and Neanderthals. A second, not mutually exclusive, possibility is that they entered the Denisova population through gene flow from some even more diverged hominin. Although such gene flow cannot be detected with the current mtDNA and nuclear DNA data, further sequencing of other hominin remains may in the future allow testing for it. A third possibility that could account for the apparently archaic dental morphology, but not the mtDNA, is a reversal to ancestral traits.
After they diverged from one another, Denisovans and Neanderthals had largely separate population histories as shown by a number of observations. First, patterns of allele sharing indicate that Denisovan ancestors did not contribute genes at a detectable level to present-day people all over Eurasia whereas Neanderthals did8. Thus, Neanderthals at some point interacted with ancestors of present-day Eurasians independently of Denisovans. Second, the genetic diversity of Neanderthals across their geographical range in the last thirty or forty thousand years of their history was extremely low, indicating that they experienced one or more strong genetic bottlenecks independently of the Denisovans. Third, our results indicate that Denisovans but not Neanderthals contributed genes to ancestors of present-day Melanesians. Fourth, the dental morphology shows no evidence of any derived features seen in Neanderthals. In fact, dental remains from the Sima de los Huesos of Atapuerca, for which ages between 350,000 and 600,000 years have been proposed39,40, already carry Neanderthal-like morphological features that are not seen in the Denisova molar.
An interesting question is how widespread Denisovans were. A possibility is that they lived in large parts of East Asia at the time when Neanderthals were present in Europe and western Asia. One observation compatible with this possibility is that Denisovan relatives seem to have contributed genes to present-day Melanesians but not to present-day populations which currently live much closer to the Altai region such as Han Chinese or Mongolians (Table 1). Thus, they have at least at some point been present in an area where they interacted with the ancestors of Melanesians and this was presumably not in southern Siberia. Further studies of both molecular and morphological features of hominin remains across Asia should clarify how widespread Denisovans were and how they were related to archaic hominins other than Neanderthals.
The Denisova individual belongs to a hominin group that shares a common ancestor with Neanderthals but has a distinct population history. We define this group based on genomic evidence and call it Denisovans, but refrain from any formal Linnaean taxonomic designations that would indicate species or subspecies status for either Neanderthals or Denisovans. In our view, these results show that on the Eurasian mainland there existed at least two forms of archaic hominins in the Upper Pleistocene: a western Eurasian form with morphological features that are commonly used to define them as Neanderthals, and an eastern form to which the Denisova individuals belong. In the future, when more complete genomes from these and other archaic hominins will be sequenced from remains that allow more morphological features to assessed, their relationships will become even better understood. This will be an important endeavour as the emerging picture of Upper Pleistocene hominin evolution is one in which gene flow among different hominin groups was common.
Methods Summary
The thirteen sections of the Supplementary Information provide a full description of the methods.
Accession codes
Primary accessions
EMBL/GenBank/DDBJ
Data deposits
The raw sequence data from the two Denisova fossils, the seven present-day humans, and the tooth mtDNA have been deposited in the European Nucleotide Archive at EMBL-EBI under accession numbers ERP000318, ERP000121 and FR695060, respectively. The alignments of Denisova sequence reads to the human and chimpanzee genomes are accessible for browsing and download from http://genome.ucsc.edu/Denisova.
References
Hublin, J. J. The origin of Neandertals. Proc. Natl Acad. Sci. USA 106, 16022–16027 (2009)
Pope, G. G. Craniofacial evidence for the origin of modern humans in China. Am. J. Phys. Anthropol. 35 (Suppl. 15). 243–298 (1992)
Rightmire, G. P. Brain size and encephalization in early to Mid-Pleistocene Homo. Am. J. Phys. Anthropol. 124, 109–123 (2004)
Wu, X. & Poirier, F. E. Human Evolution in Asia (Oxford Univ. Press, 1995)
Brown, P. et al. A new small-bodied hominin from the Late Pleistocene of Flores, Indonesia. Nature 431, 1055–1061 (2004)
Morwood, M. J. et al. Archaeology and age of a new hominin from Flores in eastern Indonesia. Nature 431, 1087–1091 (2004)
Morwood, M. J. et al. Preface: research at Liang Bua, Flores, Indonesia. J. Hum. Evol. 57, 437–449 (2009)
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010)
Beauval, C. et al. A late Neandertal femur from Les Rochers-de-Villeneuve, France. Proc. Natl Acad. Sci. USA 102, 7085–7090 (2005)
Briggs, A. W. et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325, 318–321 (2009)
Caramelli, D. et al. A highly divergent mtDNA sequence in a Neandertal individual from Italy. Curr. Biol. 16, R630–R632 (2006)
Green, R. E. et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134, 416–426 (2008)
Krause, J. et al. Neanderthals in central Asia and Siberia. Nature 449, 902–904 (2007)
Krings, M. et al. A view of Neandertal genetic diversity. Nature Genet. 26, 144–146 (2000)
Lalueza-Fox, C. et al. Mitochondrial DNA of an Iberian Neandertal suggests a population affinity with other European Neandertals. Curr. Biol. 16, R629–R630 (2006)
Orlando, L. et al. Revisiting Neandertal diversity with a 100,000 year old mtDNA sequence. Curr. Biol. 16, R400–R402 (2006)
Serre, D. et al. No evidence of Neandertal mtDNA contribution to early modern humans. PLoS Biol. 2, e57 (2004)
Endicott, P., Ho, S. Y. & Stringer, C. Using genetic evidence to evaluate four palaeoanthropological hypotheses for the timing of Neanderthal and modern human origins. J. Hum. Evol. 59, 87–95 (2010)
Krause, J. et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464, 894–897 (2010)
Derevianko, A. P. et al. Paleoenvironment and Paleolithic Human Occupation of Gorny Altai [in Russian] (Institute of Archeology and Ethnography SB RAS Press, 2003)
Derevianko, A. The Paleolithic of Siberia: New Discoveries and Interpretations (Univ. Illinois Press, 1998)
Derevianko, A. P. et al. Archaeology, Geology, and the Pleistocene and Holocene Palaeogeography of the Mountainous Altai [in Russian] (Nauka, 1998)
Derevianko, A. P., Shunkov, M. V. & Volkov, P. V. A Paleolithic bracelet from Denisova cave. Archaeol. Ethnol. Anthropol. Eurasia 34, 13–25 (2008)
Green, R. E. et al. The Neandertal genome and ancient DNA authenticity. EMBO J. 28, 2494–2502 (2009)
Rohland, N. & Hofreiter, M. Comparison and optimization of ancient DNA extraction. Biotechniques 42, 343–352 (2007)
Lindahl, T. et al. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 252, 3286–3294 (1977)
Briggs, A. W. et al. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 38, e87 (2010)
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010. 10.1101/pdb.prot5448 (2010)
Briggs, A. W. et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA 104, 14616–14621 (2007)
Kircher, M., Stenzel, U. & Kelso, J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 10, R83 (2009)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Paten, B. et al. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 18, 1814–1828 (2008)
Reich, D. et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet. 5, e1000360 (2009)
Keinan, A., Mullikin, J. C., Patterson, N. & Reich, D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nature Genet. 39, 1251–1255 (2007)
Li, J. Z. et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104 (2008)
Maricic, T., Whitten, M. & Pääbo, S. Multiplexed DNA Sequence Capture of Mitochondrial Genomes Using PCR Products. PLoS ONE 5, e14004 (2010)
Poinar, H. N. et al. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science 311, 392–394 (2006)
Rasmussen, M. et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463, 757–762 (2010)
Bischoff, J. L. et al. High-resolution U-series Dates from the Sima de los Huesos Hominids Yields 600+∞−66 kyrs: Implications for the Evolution of the Early Neanderthal Lineage. J. Archaeol. Sci. 34, 763–770 (2007)
Bischoff, J. L. et al. The Sima de los Huesos Hominids Date to Beyond U/Th Equilibrium (>350 kyr) and Perhaps to 400–500 kyr: New Radiometric Dates. J. Archaeol. Sci. 30, 275–280 (2003)
Acknowledgements
We thank C. Bustamante, A. I. Krivoshapkin, M. Lachmann, R. Nielsen, K. Pruefer, A. Tsybankov, L. Vigilant and W. Zhai for comments; K. Finstermeier for graphics work; the MPI-EVA sequencing group, R. Schultz and S. Weihnachtsmann for technical support; and P. Fujita, A. Hinrichs and K. Learned for designing the UCSC genome browser portal to the Denisova data. The Presidential Innovation Fund of the Max Planck Society and the Krekeler Foundation provided financial support. M.S. was supported by a US National Institutes of Health grant (R01-GM40282). The National Science Foundation provided an International Postdoctoral Fellowship (OISE-0754461) to J.M.G., a Fellowship in Biological Informatics to P.L.F.J. and a HOMINID grant (1032255) to D.R.
Author information
Authors and Affiliations
Contributions
J.Kr., T.M., Q.F. and M.M. performed the experiments; D.R., R.E.G., M.K., J.Kr., N.P., E.Y.D., A.W.B., U.S., P.L.F.J., T.M., J.M.G., T.B.-M., C.A., S.M., H.L., E.E.E., M.St., J.Ke., M.Sl. and S.P. analysed genetic data; B.V., M.R., S.T., M.V.S., A.P.D. and J.-J.H. analysed archaeological and anthropological data; D.R. and S.P. wrote and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains 13 Supplementary Information sections (see Table of Contents), which include Supplementary Data, Supplementary Figures, Supplementary Tables and additional references. For Supplementary Data 1 Please go to the following link to access this file: http://bioinf.eva.mpg.de/download/DenisovaGenome/Denisova_Neandertal_catalog.tgz (PDF 4630 kb)
Supplementary Data 2
This spreadsheet describes the 13 regions of potential admixture from Neandertals into modern humans as described in Green et al., Science 328:710 (2010). (XLS 60 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Reich, D., Green, R., Kircher, M. et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 (2010). https://doi.org/10.1038/nature09710
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature09710
This article is cited by
-
Dating ancient splits in phylogenetic trees, with application to the human-Neanderthal split
BMC Genomic Data (2024)
-
More than a decade of genetic research on the Denisovans
Nature Reviews Genetics (2024)
-
The Allen Ancient DNA Resource (AADR) a curated compendium of ancient human genomes
Scientific Data (2024)
-
New dating indicates intermittent human occupation of the Nwya Devu Paleolithic site on the high-altitude central Tibetan Plateau during the past 45,000 years
Science China Earth Sciences (2024)
-
Optimize or satisfice in engineering design?
Research in Engineering Design (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
