
Abstract
We report here the genome sequence of an ancient human. Obtained from ∼4,000-year-old permafrost-preserved hair, the genome represents a male individual from the first known culture to settle in Greenland. Sequenced to an average depth of 20×, we recover 79% of the diploid genome, an amount close to the practical limit of current sequencing technologies. We identify 353,151 high-confidence single-nucleotide polymorphisms (SNPs), of which 6.8% have not been reported previously. We estimate raw read contamination to be no higher than 0.8%. We use functional SNP assessment to assign possible phenotypic characteristics of the individual that belonged to a culture whose location has yielded only trace human remains. We compare the high-confidence SNPs to those of contemporary populations to find the populations most closely related to the individual. This provides evidence for a migration from Siberia into the New World some 5,500 years ago, independent of that giving rise to the modern Native Americans and Inuit.
Similar content being viewed by others

Main
Recent advances in DNA sequencing technologies have initiated an era of personal genomics. Eight human genome sequences have been reported so far, for individuals with ancestry in three distinct geographical regions: a Yoruba African1,2, four Europeans2,3,4,5, a Han Chinese6, and two Koreans7,8, and soon this data set will expand significantly as the ‘1000 genomes’ project is completed.
From an evolutionary perspective, however, modern genomics is restricted by not being able to uncover past human genetic diversity and composition directly. To access such data, ancient genomic sequencing is needed. Presently no genome from an ancient human has been published, the closest being two data sets representing a few megabases (Mb) of DNA from a single Neanderthal9,10. Contamination and DNA degradation have also compromised the possibility of obtaining high sequence depth11, and no ancient nuclear genome has been sequenced deeper than about 0.7×12—a level insufficient for genotyping and exclusion of errors owing to sequencing or post-mortem DNA damage13.
In 2008 we used permafrost-preserved hair from one of the earliest individuals that settled in the New World Arctic (northern Alaska, Canada and Greenland) belonging to the Saqqaq Culture (a component of the Arctic Small Tool tradition; approximately 4,750–2,500 14C years before present (yr bp))14,15 to generate the first complete ancient human mitochondrial DNA (mtDNA) genome16. A total of 80% of the recovered DNA was human, with no evidence of modern human contaminant DNA. Thus, the specimen is an excellent candidate upon which to sequence the first ancient human nuclear genome. Although cultural artefacts from the Arctic Small Tool tradition are found many places in the New World Arctic, few human remains have been recovered. Thus, the sequencing project described here is a direct test of the extent to which ancient genomics can contribute knowledge about now-extinct cultures, from which little is known about their phenotypic traits, genetic origin and biological relationship to present-day populations.
Sample characteristics, DNA quality and sequencing strategy
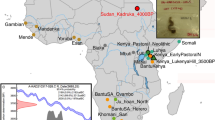
The specimen used for genomic sequencing is the largest (approximately 15 × 10 cm) of four human hair tufts excavated directly from culturally deposited permafrozen sediments at Qeqertasussuk (Fig. 1a, b). Stable light isotope analyses of the Saqqaq hair (carbon and nitrogen) revealed that the individual relied on high trophic level marine food resources (Fig. 1e and Supplementary Information). Accelerator mass spectrometry (AMS) radiocarbon dating of the hair sample produced a date of 4,044 ± 31 14C yr bp and 4,170–3,600 cal. yr bp when correcting for local marine reservoir effect (Supplementary Information). Despite its age, morphological analysis of the hair tuft using light and scanning electron microscopes indicated excellent overall preservation (Fig. 1c, d and Supplementary Information).

a, Location of the Saqqaq Culture site Qeqertasussuk, north-western Greenland (after ref. 15). b, Saqqaq hair sample. c, Saqqaq and modern hair shafts on a comparison microscope. d, Comparative cross-sections of modern Caucasian and Saqqaq hairs. e, Carbon and nitrogen isotope measurements on the Saqqaq hair (brown square, Qt 86 profile C 85/261:12 Oxford; pink triangle, Qt 86 profile C 85/261:12 Bradford), another Saqqaq hair sample from a similar context (green diamond, Qt 87 FB 20/20), six ancient Thule (Inuit) samples (purple circle), published data on modern Uummannaq (Greenland) omnivores (orange diamond), and modern Danish omnivores (blue square) and vegans (orange circle). Saqqaq and Thule are shown as averages of 2–3 replicates (Supplementary Information). Error bars, s.d. f, Calibrated ages (before present) on the Saqqaq hair and associated reindeer (Rangifer tarandus) bones, plotted using the INTCAL04 calibration curve40, are shown. The human hair dates are calibrated twice, one using a correction for the marine reservoir effect (Supplementary Information).
A major concern in ancient DNA studies is post-mortem damage, cytosine to uracil deamination, that can result in erroneous base incorporation17,18. Such miscoding lesions make it difficult to distinguish true evolutionarily derived substitutions from those that are damage-based, especially if sequence depth is low. It is therefore preferential to exclude damaged DNA molecules before sequencing, if achievable without loss of significant amounts of starting templates. We established the practical feasibility of this, by comparing Illumina sequencing libraries that were initially enriched using two different DNA polymerase enzymes: (1) Phusion polymerase (Finnzymes) as suggested in Illumina’s own library preparation protocol, which is not able to replicate through uracil19; and (2) Platinum Taq High Fidelity (HiFi, Invitrogen) polymerase, that can replicate through uracil (Supplementary Information).
Results allowed us to estimate an overall deamination-based damage rate in the Saqqaq genome of <1%, which is, as expected, lower than the rate obtained from GS FLX sequencing16 (Supplementary Information). We also found undamaged sequences to be slightly shorter on average than those containing damage (55 base pairs (bp) for Phusion versus 59 bp for HiFi). However, given that GS FLX shotgun sequencing shows an average molecular length of <76 bp in the Saqqaq hair sample16 (a known overestimate due to automatic computational filtering of short reads), and that quantitative polymerase chain reaction (qPCR) revealed high copy numbers of short fragments (approximately 1.8 million copies per microlitre DNA extract of 85-bp mtDNA), dropping roughly exponentially with sequence length (Supplementary Fig. 10), we concluded that excluding damaged molecules makes little difference to the number of starting DNA molecules available for initial sequence enrichment.
Ancient human DNA is particularly susceptible to contamination by modern DNA20. Although the qPCR results confirmed that DNA preservation in the Saqqaq hair is excellent as judged by ancient DNA standards21, we undertook several actions before sequencing to minimize and control for contamination. In addition to using a decontamination protocol that has previously proven successful on the Saqqaq hair sample16, we also used indexing adaptors and primers in the library preparations13, such that any possible contamination entering the samples after they left the ancient DNA clean laboratory in Copenhagen could be easily detected (Supplementary Information). This ensures that any possible human contamination should reveal itself as being of European origin, given that any handling steps before indexing were carried out only by ethnic northern Europeans (Supplementary Information).
Sequencing and assembly
Twelve DNA libraries were built in the dedicated Copenhagen ancient DNA laboratory, several indexed enrichment PCRs were carried out, and each was sequenced on an average of three lanes using Illumina GAII sequencing platforms at BGI-Shenzhen. In addition, two sequencing runs were completed at Illumina’s facilities in Hayward, California and Chesterford, England. With few exceptions, 70 cycles of single-read sequencing were performed, always followed by a 6-bp indexing read (Supplementary Information). The sequencing yielded a total of 3.5 billion reads, from a total of 242 lanes.
Sequences not carrying a 100% match in the index read were excluded from all downstream analyses. This allowed 93.17% of all reads to be attempted to be mapped to the human reference genome (hg18) using a suffix array-based mapping strategy that permits identification of residual primer sequence expected from the libraries of short ancient DNA fragments (Supplementary Information). Primer trimming was carried out as an integrated part of the mapping during the alignment of each read to the genome. Specifically, for all positions a check was made as to whether a better alignment could be made between the remainder of the read and the primer. If found, this position in the read was used to cut off the primer (Supplementary Information). This provided an average mapped read length of 55.27 nucleotides. Of the correctly indexed reads, 49.2% could be mapped uniquely (46% of total reads). Reads with multiple matches or no matches were discarded (Fig. 2a). Analysis of the reads with no matches indicated that most were unidentifiable, whereas the remainder were of microbial eukaryote, viral, or bacterial origin (Fig. 2b). Read sequences from the same library that were mapped to the reference genome with same start and end positions were considered clonal, and were collapsed to single sequences with higher quality scores (Supplementary Information). This resulted in a final data set of 28.47% of all reads. Additionally, to avoid erroneous SNP calls due to insertions and deletions, we discarded the last seven nucleotides from the 3′ end of the mapped reads, yielding a final average read size of 48 nucleotides. This provides an average depth of 20× across 79% of the genome (Fig. 2a). Given a maximum read length of 70 bp and an average mapped read length of 55 bp, we estimate that it is theoretically possible to cover some 85–87% of the genome (Supplementary Information), meaning that we are close to having sequenced all that is feasible with the technology at hand. Approximately one-half of the positions are covered with a depth >7×, with some variation along the chromosomes, largely explained by repetitive structures in the genome, which can both artificially raise or lower the depth locally (Fig. 2c–f).

a, Flow diagram summarizing our data pipeline. b, Distribution of all reads among the major taxonomic groups. c, Cumulative distribution of reads across the genome, for all positions (black), only repeat regions (blue) or exclusive repeat regions (green). d, The read depth (green) varies along the chromosomes. e, Much of this variation can be attributed to the repetitive structure of the genome and is especially pronounced in highly repetitive regions, for example, flanking the centromere, but is also observed in regions with genes (shades of blue). f, Simple repeats, here (CAGC) n (grey), are common in assembly gaps and therefore cause alleviated read depths. In contrast, a recent segmental duplication (black, with differences in red) will prevent reads from mapping uniquely and lower the read depth.
Genotyping and comparative genomic analyses
For genotyping, we developed a probabilistic model of the sampling of reads from the diploid genome, called SNPest, which takes quality scores and different sources of read errors into account. For the sex chromosomes and the mtDNA a haploid model was used. Given the mapped reads and their quality scores, we assigned the most probable genotype to each position (Supplementary Information). We performed genotyping on all positions, using all available read information for depths ≤200×. For read depths >200×, we based the genotyping on 200 randomly sampled reads. This simplification was shown to have negligible effect on the results while speeding up the calculations markedly (Supplementary Information). This resulted in 2.2 million SNPs (Fig. 2a), of which 86.2% have previously been reported (dbSNPv130).
We additionally defined a high-quality subset of SNPs, based on positions with read depth between 10× and 50×, to avoid poorly covered and repetitive regions with extreme read depth. We also demanded that these SNPs have posterior probabilities of >0.9999, not to be positioned in annotated repeat regions, and to have a distance of at least 5 bp to the closest neighbouring SNP to account for insertion and/or deletion (indel) errors6. This provided a total of 353,151 SNPs with a 93.2% overlap with dbSNP (v130) (Fig. 2a).
The mtDNA genome was sequenced to an average depth of 3,802×. The consensus was identical to that previously recovered by GS FLX sequencing, except that a single position previously called as a heterozygote16 was now called as a C. Using the diploid model, no high-confidence heterozygotes were found. Applying the diploid model to the X chromosome resulted in 1,707 homozygote (versus 3,071 with the haploid model) and 76 heterozygote high-confidence SNPs. Of the latter, 29% can be explained by known indels and structural variation, whereas the remaining can be referred to mapping errors in repetitive regions (Supplementary Information). For the Saqqaq Y chromosome, we found 23 homozygote (versus 243 with the haploid model) and 445 heterozygote high-confidence SNPs. We explain the latter by the well-known fact that human Y chromosomes are difficult to assemble due to structural and repetitive regions22. Importantly, the number of heterozygote SNPs found in the X and Y chromosomes when changing to the diploid model are similar to those from modern human genome sequencing (Supplementary Information).
Assessing contamination using the frequency of private European alleles (as defined in the human genome diversity project) as an estimator and a fixed error rate from the observed neighbouring bases, we estimate the raw read contamination to be at most 0.8% (standard error (s.e.) ± 0.2%) (Supplementary Information), a level that will not affect our high-confidence genotype calls and will have a negligible effect otherwise.
We investigated the Saqqaq individual for signs of inbreeding using two new statistical approaches that circumvent the problem of uncertainty in the genotype calls of heterozygotes, using the Siberian populations from Supplementary Table 12 as a reference. The methods provide a genome-wide estimate of the inbreeding coefficient (F) and identify regions of identity by descent (IBD) across the genome (Supplementary Fig. 13). The estimated value of F is 0.06 (s.e. 0.011) assuming no genotyping errors, which is equivalent to an offspring of two first cousins, but could have been caused by other family relationships of the parents (Supplementary Information). A positive value of F could possibly also be explained by population subdivision between the Saqqaq population and the Siberian reference population, or by natural selection. However, as many IBD tracts are >10 Mb, far longer than the extent of linkage disequilibrium in the human genome, inbreeding within the Saqqaq population is more likely.
Functional SNP assessment
Although the relationship between risk allele and causation is still in its infancy23, some phenotypic traits can possibly be inferred from the genome data (all functional SNPs discussed below are listed in Supplementary Table 14). We only included genotypes with a posterior probability above 99%.
Given the A1 antigen allele plus encoding of the rhesus factor in combination with lack of B antigen and the O antigen frameshift mutation, we conclude that the Saqqaq individual had blood type A+24. Although common in all ethnic groups, this has very high frequencies in populations of the east coast of Siberia down to mid China25. Furthermore, we find a combination of four SNPs at the HERC2-OCA2 locus, which among Asians is strongly associated with brown eyes26. SNPs on chromosomes 2, 5, 15 and X suggest that he probably did not have a European light skin colour27, had dark and thick hair28,29 (in agreement with the morphological examination (Fig. 1b–d)), and an increased risk of baldness30,31. The same SNP that is characteristic of hair thickness also suggests that he probably had shovel-graded front teeth—a characteristic trait of Asian and Native American populations32. An AA genotype SNP (forward strand) on chromosome 16 is consistent with the Saqqaq individual having earwax of the dry type that is typical of Asians and Native Americans, rather than the wet earwax type dominant in other ethnic groups33. In addition, the combined influence of 12 SNPs on metabolism and body mass index indicates that the Saqqaq individual was adapted to a cold climate (see Supplementary Information and Supplementary Table 14).
Population genetics context of the Saqqaq individual
The origin of the Saqqaq and other Palaeo-Eskimo cultures, and their relationship to present-day populations, has been debated since they were first discovered in the 1950s34. Competing theories have attributed the origins to offshoots of the populations that gave rise to Native American populations such as the Na-Dene of North America, alternatively from the same source as the Inuit currently inhabiting the New World Arctic, or from still other sources entering the New World even later than both the Native American and Inuit ancestors (for summary see ref. 35).
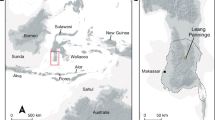
A recent SNP genotyping study36 of the HGDP-CEPH panel of 51 populations has provided comprehensive global coverage of modern human genomic variation, but is limited with respect to Arctic populations. Therefore, we carried out Illumina Bead-Array-based genotyping on four native North American and twelve north Asian populations (Supplementary Table 12). A total of 95,502 SNPs from the resulting combined data set of 35 Eurasian and American populations was covered by high-quality data in the Saqqaq genome and was subject to further analyses (Fig. 3a–c and below).

a, Locations of the studied populations are shown with the most relevant populations indicated by name (numbers in circles correspond to the nr column in Supplementary Table 12). b, PCA plot (PC1 versus PC2) of the studied populations and the Saqqaq genome. c, Ancestry proportions of the studied 492 individuals from 35 extant American and Eurasian populations and the Saqqaq individual as revealed by the ADMIXTURE program39 with K = 5. Each individual is represented by a stacked column of the five proportions, with fractions indicated on the y axis. The analysis assumes no grouping information. The samples are sorted by region/population only after the analysis. For better readability the Saqqaq individual is shown in three columns. Populations added to the published collection36 are shown in bold. Red dots in the expanded plot indicate four individuals whose ancestry proportion pattern showed the highest correlation (Kendall τ > 0.95; P < 0.05) with that of the Saqqaq individual. d, The phylogenetic tree of Y chromosome haplogroup Q. The position of the Saqqaq individual is ascertained by markers shown on the tree. Information for markers shown in parentheses is missing and their status is therefore inferred. Haplogroup names are according to ref. 38; hash symbol indicates error in reference (Supplementary Information).
Principal component analysis (PCA) was used to capture genetic variation. PC1 distinguishes west Eurasians from east Asians and Native Americans, whereas the PC2 captures differentiation between native Asians and Americans (Fig. 3b). Importantly, the PC1 versus PC2 plot shows that the Saqqaq individual falls in the vicinity of three Old World Arctic populations—Nganasans, Koryaks and Chukchis, while being more distantly related to the New World groups (Amerinds, Na-Dene and Greenland Inuit). Koryaks and Chukchis inhabit Chukotka and northern Kamchatka of the Siberian far east. Ethnography describes these groups as having a diverse subsistence economy based on terrestrial and marine hunting as well as reindeer herding. The Nganasans inhabit the Taimyr Peninsula, some 2,000 km from the Bering Strait and are the northernmost living Old World population. Although historically Nganasans have been terrestrial rather than marine hunters, Zhokov, the oldest archaeological Arctic hunting site with a significant marine component (polar bear) on the New Siberian Islands (dating back some 7,000–8,000 yr bp37), is found just east of the Nganasans’ current occupation area. In addition, our analysis of more than two hundred Y chromosome SNPs (Supplementary Information) allowed us to assign the Saqqaq individual to Y chromosome haplogroup Q1a (Fig. 3d), commonly found among Siberian and Native American populations38. The mtDNA genome shows close relatedness to Aleuts of Commander Islands (situated in the Bering Sea) and Siberian Sireniki Yuits (Asian Eskimos) as previously described16.
We explored the data using the algorithm ADMIXTURE39, which assumes a specified number of hypothetical populations (K) and provides a maximum likelihood estimate of allele frequencies for each population and admixture proportion for each individual. We investigated values of K, from K = 2 to K = 10, repeating computing 100 times for each value of K to monitor convergence (Supplementary Information). Figure 3c shows the pattern of distinct colour-coded components at K = 5. The analysis suggests that there is a significant amount of west Eurasian admixture in most of the Siberian, Greenland and North American populations. As with the other analyses, this analysis was unable to detect any west Eurasian admixture in the Saqqaq individual, in agreement with a very low level of contamination in our assembled genome. The Saqqaq individual is also practically devoid of the component distinctive to South and Central American populations (dark brown in Fig. 3c). Thus, at K = 5, the Saqqaq genome is comprised of three ethnic influences, specifically the ones characteristic of native populations in East Asia, Siberia in particular, and the Arctic, on both sides of the Bering Strait (Fig. 3c). In this respect the populations closest to the Saqqaq are Koryaks and Chukchis. Importantly, in contrast to Saqqaq and Koryaks, modern Greenlanders carry clear evidence of admixture or shared ancestry with Amerindians. Moreover, at K = 5, the Inuit do not display genetic components of Siberians other than the ‘Beringian’ seen in Chukchis and Koryaks. The admixture results are in agreement with the PCA plots and suggest shared common ancestry of Saqqaq and modern Inuit before the movement of the former to the New World.
We additionally used a population genetic model to obtain maximum likelihood estimates of the divergence times between the Saqqaq individual and the reference populations (Supplementary Information). The population with the shortest divergence time was Chukchis, with an estimated divergence time of approximately 0.043 (±0.08) Ne generations, where Ne is the effective population size. In contrast, the estimated divergence times to the other closely related populations—Na-Dene, Koryaks and Nganasans—were 0.093, 0.11 and 0.089, respectively. The estimated divergence time to the Han Chinese, a more distantly related population, was 0.20. These estimates can be converted to estimates of years or generations, by making assumptions regarding the effective population sizes of the reference populations. The effective population sizes are in general unknown, but can be estimated from DNA sequence data, and are generally much smaller than the census sizes (Supplementary Information). We found no evidence in favour of changes in population size. Even when accounting for the uncertainty in the estimate of the mtDNA mutation rate, and possible biases related to the genotyping data, it is still unlikely that Ne > 5,000, providing a maximal divergence time between Chukchis and Saqqaqs of 175–255 generations or between 4,400 and 6,400 years. The oldest archaeological evidence of the Arctic Small Tool tradition in the New World is from Kuzitrin Lake, Alaska, dating back ∼5,500 cal. yr bp14, indicating that the ancestral Saqqaq separated from their Old World relatives almost immediately before their migration into the New World.
Conclusion
We report the successful genome sequencing of a ∼4,000-year-old human. Data authenticity is supported by: (1) the private SNP analyses that indicate contamination levels in the raw sequence data to be ≤0.8%; (2) the mtDNA and Y-chromosome DNA haplotypes fit within haplogroups typical of north-east Asia; (3) population admixture analyses do not record any European component in the Saqqaq genome; and (4) the PCA plots clearly reveal close affiliation of the Saqqaq genome to those of contemporary north-east Siberian populations. These observations, coupled with evidence of excellent DNA preservation, and sample handling being restricted to northern Europeans before incorporation of a sequence indexing, indicate that contamination in the Saqqaq genome is not of concern. Our study thus demonstrates that it is possible to sequence the genome of an ancient human to a level that allows for SNP and population analyses to take place. It also reveals that such genomic data can be used to identify important phenotypic traits of an individual from an extinct culture that left only minor morphological information behind. Additionally, the ancient genomic data prove important in addressing past demographic history by unambiguously showing close relationship between Saqqaq and Old World Arctic populations (Nganasans, Koryaks and Chukchis). A single individual may, or may not, be representative of the extinct culture that inhabited Greenland some 4,000 yr bp. Nevertheless, we may conclude that he, and perhaps the group that once crossed the Bering Strait, did this independently from the ancestors of present-day Native Americans and Inuit, and that he shares ancestry with Arctic north-east Asians, genetic structure components of which can be identified in many of the present-day people on both sides of the Bering Sea. The next technical challenge will be to sequence an ancient human genome from material outside the permafrost regions. Although undoubtedly challenging, it will, if successful, take the emerging field of palaeogenomics to yet another level.
Methods Summary
DNA was extracted from a ∼4,000-year-old hair sample recovered from Qeqertasussuk, Greenland. Indexed Illumina libraries were sequenced following the manufacturer’s protocol, and images processed using pipeline v1.4. Reads with correct index were mapped to the human genome (hg18) with a suffix array-based method that allows for residual primer trimming (Supplementary Information). Genotyping was carried out using a probabilistic model, SNPest, designed to take into account errors specific for ancient samples (Supplementary Information).
Accession codes
Data deposits
Sequences have been deposited to the short read archive with accession number SRA010102; summary data are also available via http://www.ancientgenome.dk.
References
Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008)
Drmanac, R. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81 (2010)
Levy, S. et al. The diploid genome sequence of an individual human. PLoS Biol. 5, e254 (2007)
Wheeler, D. A. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–876 (2008)
Pushkarev, D. et al. Single-molecule sequencing of an individual human genome. Nature Biotechnol. 27, 847–850 (2009)
Wang, J. et al. The diploid genome sequence of an Asian individual. Nature 456, 60–65 (2008)
Ahn, S.-M. et al. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 19, 1622–1629 (2009)
Kim, J.-I. et al. A highly annotated whole-genome sequence of a Korean individual. Nature 460, 1011–1015 (2009)
Noonan, J. P. et al. Sequencing and analysis of neanderthal genomic DNA. Science 314, 1113–1118 (2006)
Green, R. E. et al. Analysis of one million base pairs of Neanderthal DNA. Nature 444, 330–336 (2006)
Wall, J. D. & Kim, S. K. Inconsistencies in neanderthal genomic DNA sequences. PLoS Genet. 3, 1862–1866 (2007)
Miller, W. et al. Sequencing the nuclear genome of the extinct woolly mammoth. Nature 456, 387–390 (2008)
Green, R. E. et al. The neandertal genome and ancient DNA authenticity. EMBO J. 28, 2494–2502 (2009)
Harritt, R. Paleo-eskimo beginnings in North America: a new discovery at Kuzitrin lake, Alaska. Etud. Inuit 22, 61–81 (1998)
Meldgaard, M. Ancient Harp Seal Hunters of Disko Bay. Subsistence and Settlement at the Saqqaq Culture Site Qeqertasussuk (2400–1400 BC), West Greenland. Meddelelser om Grønland, Man & Society (Danish Polar Center, 2004)
Gilbert, M. T. P. et al. Paleo-eskimo mtDNA genome reveals matrilineal discontinuity in Greenland. Science 320, 1787–1789 (2008)
Pääbo, S. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl Acad. Sci. USA 86, 1939–1943 (1989)
Brotherton, P. et al. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 35, 5717–5728 (2007)
Fogg, M. J. et al. Structural basis for uracil recognition by archaeal family b DNA polymerases. Nature Struct. Biol. 9, 922–927 (2002)
Willerslev, E. & Cooper, A. Ancient DNA. Proc. Biol. Sci. 272, 3–16 (2005)
Handt, O. et al. The retrieval of ancient human DNA sequences. Am. J. Hum. Genet. 59, 368–376 (1996)
Skaletsky, H. et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003)
Benfey, P. N. & Mitchell-Olds, T. From genotype to phenotype: systems biology meets natural variation. Science 320, 495–497 (2008)
Yamamoto, F. et al. Molecular genetic basis of the histo-blood group ABO system. Nature 345, 229–233 (1990)
Cavalli-Sforza, L. L. et al. The History and Geography of Human Genes (Princeton Univ. Press, 1994)
Iida, R. et al. Genotyping of five single nucleotide polymorphisms in the OCA2 and HERC2 genes associated with blue-brown eye color in the Japanese population. Cell Biochem. Funct. 27, 323–327 (2009)
Soejima, M. & Koda, Y. Population differences of two coding SNPs in pigmentation-related genes SLC24A5 and SLC45A2 . Int. J. Legal Med. 121, 36–39 (2007)
Branicki, W. et al. Association of the SLC45A2 gene with physiological human hair colour variation. J. Hum. Genet. 53, 966–971 (2008)
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007)
Prodi, D. A. et al. EDA2R is associated with androgenetic alopecia. J. Invest. Dermatol. 128, 2268–2270 (2008)
Ellis, J. A. et al. Baldness and the androgen receptor: the AR polyglycine repeat polymorphism does not confer susceptibility to androgenetic alopecia. Hum. Genet. 121, 451–457 (2007)
Kimura, R. et al. A common variation in EDAR is a genetic determinant of shovel-shaped incisors. Am. J. Hum. Genet. 85, 528–535 (2009)
Yoshiura, K. et al. A SNP in the ABCC11 gene is the determinant of human earwax type. Nature Genet. 38, 324–330 (2006)
Meldgaard, J. A. Paleo-Eskimo culture in West Greenland. Am. Antiq. 17, 222–230 (1952)
McGhee, R. Canadian Arctic Prehistory. Canadian Prehistory Series (Canadian Museum of Civilization, 1990)
Li, J. Z. et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104 (2008)
Pitulko, V. & Makeyev, V. Ancient Arctic Hunters. Nature 349, 374 (1991)
Karafet, T. et al. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 18, 830–838 (2008)
Alexander, D. H. et al. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009)
Reimer, P. et al. IntCal04 terrestrial radiocarbon age calibration, 0–26 cal kyr BP. Radiocarbon 46, 1029–1058 (2004)
Acknowledgements
Centre for Geogenetics, the Copenhagen branch of the Sino-Danish Genomic Centre and Wilhelm Johannsen Centre for Functional Genome Research were supported by Danish National Research Foundation, the Lundbeck Foundation, and the Danish Agency for Science, Technology and Innovation. Center for Biological Sequence Analysis was supported by Villum Kann Rasmussen Fonden; Center for Protein Reseaerch by the Novo Nordisk Foundation. E.W. thanks F. Paulsen for financial support to initiate the project. E.M. thanks Estonian Science Foundation for grant 7858, and R.V. EC DGR for FP7 Ecogene grant 205419 and EU RDF through Centre of Excellence in Genomics grant. J.W. thanks the Shenzhen Municipal Government, the Yantian District local government of Shenzhen, the National Natural Science Foundation of China (30725008), Ole Romer grant from the Danish Natural Science Research Council, the Solexa project (272-07-0196), and Danish Strategic Research Council (2106-07-0021). M.Bu. acknowledges the support of the Australian Research Council. A.K., S.L. and H.M.K. were supported by a grant from the Novo Nordisk Foundation and J.S.P. The Danish Council for Independent Research Medical Sciences. M.H.C. thanks the National Science Foundation for support of the Aleutian and Siberian projects through grants NSF OPP-990590 and OPP-0327676. We thank G. Hudjashov, M. Nelis, L. Anton, V. Soo, A. Wesolowska, H.-H. Staerfeldt, K. Rapacki, P. Wad Sackett, J. Li, H. Yu, Y. Huang, H. Zheng, H. Liang and T. Brand for technical help and Biobase for curation of selected phenotypes and Illumina’s core facilities in Hayward, California and Chesterford, England. Use of HGMD Professional was licensed through DTV at the Technical University of Denmark.
Author Contributions E.W. initially conceived and headed the project (J.W. headed research at BGI). M. Rasmussen and E.W. designed the experimental research project setup. T.L.P., M.Mel., C.A., B.G., S.A.F., L.P.O., M.H.C., F.C.N. and R.V. provided samples and/or modern DNA extracts. R.V. carried out Illumina chip analyses on modern populations. T.F.G.H. and C.B.R did the AMS dating. A.S.W., A.G. and S.T. did the morphological analyses. M. Raghavan, A.S.W. and A.G. did the isotope analyses. M.T.P.G., M.Bu. and M.Ras. did ancient DNA extractions. M.Ras. did the library building and qPCR. M.Ras. and P.F.C. did polymerases analyses; ancient extracts were provided by E.D.L. and J.B. Y.L., J.Z., X.G., X.Z., H.Z., Z.L., M.C. and J.W. did the Illumina sequencing and the basic analysis for the sequence raw data. S.L., J.S.P., H.M.K. and A.K. did the method development and data analysis for the genome assembly and genotyping with input from M.Ras., T.S.-P., R.G., M.Be., K.N. and S.B. did the metagenomics, genomic phylogeny and the functional SNP assignment. A.A., I.M., Y.W. and R.N. did the PCA analysis (with input from R.V.), inbreeding estimates and maximum likelihood estimates of the divergence times. M.Met., E.M. and R.V. did the admixture analyses. T.K. did the Y-chromosome analyses. E.W. and J.W. each paid half the genome sequencing costs. E.W. paid the Illumina chip analyses. E.W. and M.Ras. wrote the majority of the manuscript, with critical input from A.K., S.L., J.S.P., R.V., T.K., M.Met., R.N., I.M., A.A., M.T.P.G., T.S.-P., Y.L., J.W. and the remaining authors.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Information, Supplementary References, Supplementary Tables S1-S14 and Supplementary Figures S1-S16 with Legends. (PDF 3454 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Rasmussen, M., Li, Y., Lindgreen, S. et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463, 757–762 (2010). https://doi.org/10.1038/nature08835
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature08835
This article is cited by
-
The Allen Ancient DNA Resource (AADR) a curated compendium of ancient human genomes
Scientific Data (2024)
-
‘Truly gobsmacked’: Ancient-human genome count surpasses 10,000
Nature (2023)
-
Ancient DNA reveals genetic admixture in China during tiger evolution
Nature Ecology & Evolution (2023)
-
A common founder effect of the splice site variant c.-23 + 1G > A in GJB2 gene causing autosomal recessive deafness 1A (DFNB1A) in Eurasia
Human Genetics (2022)
-
Whole-exome sequencing of the mummified remains of Cangrande della Scala (1291–1329 CE) indicates the first known case of late-onset Pompe disease
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
