
Abstract
The engineering of materials that can modulate the immune system is an emerging field that is developing alongside immunology. For therapeutic ends such as vaccine development, materials are now being engineered to deliver antigens through specific intracellular pathways, allowing better control of the way in which antigens are presented to one of the key types of immune cell, T cells. Materials are also being designed as adjuvants, to mimic specific 'danger' signals in order to manipulate the resultant cytokine environment, which influences how antigens are interpreted by T cells. In addition to offering the potential for medical advances, immunomodulatory materials can form well-defined model systems, helping to provide new insight into basic immunobiology.
Similar content being viewed by others
Main
The term 'immunobioengineering' is used to describe efforts by immunologists and engineers to design materials, delivery vehicles and molecules both to manipulate and to better understand the immune system. Examples are the engineering of material surfaces to induce or prevent complement activation, the engineering of adjuvants to activate the immune system, the engineering of antigen or adjuvant carriers for subunit vaccine delivery, and the engineering of microenvironments to determine the interaction kinetics of mature dendritic cells and naive T cells. These advances not only will contribute to prophylactic vaccine strategies for infectious diseases but also are likely to affect immunotherapeutics, particularly for cancer, and new approaches to prevent or treat allergies and autoimmune diseases. The field is rapidly evolving along with advances in our understanding of immunology and is also contributing to our knowledge of basic immunology.
In this Review, we describe the current state of immunobioengineering as it intersects with the field of materials science, and we give a perspective on its current and future directions. We focus on materials for immunomodulation, particularly with respect to dendritic-cell modulation. We begin by providing a brief introduction to the targets for delivery: the types of cell that materials are being designed to target, the tissues in which those cells reside, the intracellular compartments within those cells, and the influence that delivery to those particular compartments has on immunological outcome. We then discuss the biomolecular payloads used to activate immune cells and the design of the materials used to deliver those 'danger' signals along with, in the context of vaccination, antigens. Finally, we highlight materials approaches that are being developed to explore basic immunological function, especially how dendritic cells and T cells interact.
Tissue and cellular targets
As the general goals of immunobioengineering are to probe and manipulate the immune system, we start with a general discussion of tissue, cellular and subcellular targets — what we want to target and why — to guide the design principles discussed below.
The immune cells most frequently targeted include B cells, macrophages and dendritic cells, which are all effective antigen-presenting cells (APCs)1. Dendritic cells are typically considered the most specialized, because they present antigen to their cognate naive T-cell partners and instruct them what do with it (for example induce anergy, tolerance or immunity)2. They are the main focus of this Review, although recent evidence points to basophils being an important APC involved in T helper 2 (TH2)-cell immunity3.
In the most simplistic, classic view, dendritic cells remain in an immature state while sampling antigens in their environment to present to T cells for the maintenance of self-tolerance (that is, they present antigen without costimulatory molecules); this includes in the lymph node, where functionally immature dendritic cells sample antigens drained with lymph from the periphery4. When they encounter pathogenic or endogenous danger signals (described below) or adjuvants (as engineered danger signals) while taking up antigen, they begin to mature and express the chemokine receptor CCR7, which allows them to migrate into the nearest draining lymphatic vessels and then to the lymph node5. There, they present processed antigenic peptides with maturation-induced costimulatory molecules to T cells to initiate an adaptive, or antigen-specific, immune response.
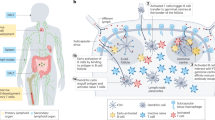
Thus, in this simplistic view, antigen presented by immature dendritic cells (in the presence of transforming growth factor-β1 (TGF-β1) and interleukin-10 (IL-10)) maintains tolerogenic responses (Fig. 1a), whereas that presented by mature dendritic cells in the presence of immunogenic cytokines can lead to immunogenic T cells. In reality, there are exceptions: mature dendritic cells can induce tolerogenic responses, and there are 'partially mature' dendritic cells, whose function is poorly understood1.

To activate naive CD4+ (helper) T cells, the T-cell antigen receptor needs to recognize antigen that has been loaded onto MHC class II molecules and presented by dendritic cells. The response of the T cells depends not only on this receptor–ligand interaction but also on costimulatory molecules being presented by the dendritic cells and on the cytokine environment during activation. a, In normal surveillance mode, dendritic cells in their immature state constantly pick up antigen and present it, without costimulatory molecules, to T cells. This leads to T-cell anergy (that is, the T cell cannot receive further signals) and/or activation of inducible (FOXP3+) regulatory T (Treg) cells when the cytokines TGF-β1 and IL-10 are present. Treg cells themselves secrete TGF-β1 and IL-10, which inhibit TH1 cells. In fact, all types of T cell can secrete IL-10. b, By contrast, when dendritic cells that have been activated by pattern-recognition receptor (PRR) ligation and maturation signals from microbial products present antigen to T cells together with costimulatory molecules, they can drive a TH1-, TH2- or TH17-cell response. For example, at the early stages of bacterial or viral infection, antigen presentation occurs in a microenvironment that contains IL-6 and IL-23 (but no IL-4), stimulating naive CD4+ T cells to differentiate into TH17 cells. These cells secrete IL-6, IL-22 (which induces epithelial cells to produce antimicrobial peptides) and IL-17 (which activates local fibroblasts). The fibroblasts, in turn, attract neutrophils and macrophages through the secretion of cytokines such as GM-CSF and IL-6 and chemokines such as CXCL8 and CXCL12. c, At later stages of infection, these activated neutrophils and macrophages, in turn, secrete the chemokines CCL3, CCL4 and CCL5, which attract additional T cells and promote the differentiation of CD4+ T cells into TH1 cells. A TH1-cell response occurs when dendritic cells mature in the presence of cytokines such as TNF-α and GM-CSF and when PRRs such as Toll-like receptors (TLRs) are activated by microbial products, leading to IL-12 secretion. IL-12 is a key cytokine for TH1-cell activation, which can be enhanced by IFN-γ. TH1 cells secrete IFN-γ, which (in addition to promoting further TH1-cell activation) activates natural killer (NK) cells and inhibits the activation of TH2 and TH17 cells. TH1 cells also produce TNF-α, as well as IL-2 and lymphotoxin-α, all of which can drive B cells to differentiate into opsonizing-antibody-producing plasma cells (which predominantly produce IgG). Under certain conditions, IL-10 is secreted by TH1 cells as an inhibitory feedback regulator. d, TH2-cell responses are elicited when dendritic cells present antigen in the presence of IL-4, which is secreted by TH2 cells themselves and inhibits TH17 cells. TH2 cells also secrete IL-5, which together with IL-4 stimulates B-cell proliferation and antibody production (especially antibody of the classes IgM, IgA and IgE). Other cytokines secreted by TH2 cells include IL-6, IL-9, IL-13 and IL-10, the last of which inhibits IFN-γ production by TH1 cells and IL-12 production by dendritic cells. Therefore, when engineering immune responses, it is important to consider the cytokine microenvironment, in addition to how the antigen will be presented by dendritic cells, both of which can be modulated by PRR signalling and uptake mechanisms.
Moreover, there are many subsets of dendritic cells, including the following: plasmacytoid dendritic cells, which secrete interferon-α (IFN-α); myeloid dendritic cells, which can secrete large amounts of IL-12; follicular dendritic cells, which do not express major histocompatibility complex (MHC) class II molecules; lymphoid dendritic cells, which can secrete large amounts of IFN-γ; CD8− or CD8+ dendritic cells, of which the former do not carry the CD8 antigen and the latter do; and several tissue-specific subtypes such as Langerhans cells. Although still poorly understood, these subtypes can be specialized for different functions, such as antigen 'cross-presentation' by CD8+DEC205+ dendritic cells (DEC205 also known as LY75) residing in the splenic T-cell zone, or MHC class II presentation by CD8− dendritic cells residing in the red pulp6. Much of what we know about dendritic-cell behaviour comes from in vitro studies using dendritic cells derived from peripheral blood monocytes, haematopoietic progenitor cells or bone marrow; these cells are typically differentiated into immature dendritic cells by using IL-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF, also known as CSF2) and matured by using lipopolysaccharide (LPS) or tumour-necrosis factor-α (TNF-α). Therefore, care must be taken in translating in vitro data from generic 'dendritic cells' to the in vivo situation, with an appreciation for tissue-specific dendritic-cell subsets, and it should be noted that there are many differences between such subsets in rodents and those in humans.
The response that a dendritic cell elicits depends on many factors, including the state of maturation of the cell, how the antigen was taken up and processed by the cell, and even the tissue in which the cell was activated. Antigens presented in the context of MHC class I molecules are recognized only by CD8+ T cells, whereas those bound to MHC class II molecules are recognized by CD4+ T cells. Dendritic cells, classically CD8+ dendritic cells, can present antigen in the context of both classes of MHC molecule. Some of these complex interactions with CD4+ T cells are illustrated in Fig. 1 and described in Table 1. Importantly, the cytokine environment in which both dendritic-cell activation and communication between dendritic cells and CD4+ T cells occurs can control the response and should be considered when choosing tissue targets. These cytokines can be secreted by dendritic cells on activation or inactivation, by the activated CD4+ T cells themselves, by neutrophils and macrophages recruited to the inflammatory site and, finally, by stromal cells that can become activated during inflammation. Because the cytokine profile present during contact with the dendritic cell can determine CD4+ T-cell fate, engineering approaches that manipulate or make use of the cytokine environment are important design considerations. For example, a biomaterials vaccine platform in which IL-10 expression was knocked down using short interfering RNA has been explored, showing strongly positive effects; from Fig. 1 and Table 1, it is evident that this was done to block the inhibitory role that IL-10 has in TH1-cell and TH2-cell development7.
APCs traffic through almost every tissue in the body. Generally speaking, epithelial tissues have strong immunosurveillance activity, as they form barriers to the outside world through which most pathogenic entry occurs. In peripheral tissues such as the skin, Langerhans cells, other dendritic cells and macrophages detect danger signal and antigens, signal other immune cells to the site by means of chemokine secretion and migrate to the nearest draining lymph node to activate an immune response. Most traditional vaccines delivered in the skin or muscle target such cells, including those vaccines formulated in alum (the standard adjuvant, which consists of particulate aluminium phosphates; described further below), an adjuvant that targets peripheral APCs through its 'depot effect'. The skin, lungs, gut and lymph nodes are common target tissues for both natural immunomodulatory agents and prophylactic or therapeutic ones, and different immune responses can be achieved in different target tissues8.
The lymph node is an emerging target tissue of interest. Dendritic cells and B cells reside in the lymph nodes, and T cells traffic through the paracortical region, the architecture of which is optimized for rapid cell trafficking to help naive T cells to make contact with thousands of dendritic cells to find their APC partners9. Immature dendritic cells patrol peripheral tissues and then migrate to the lymph nodes after they take up antigen, but the lymph nodes also contain many immature dendritic cells that constantly sample antigens carried through the lymph nodes by lymph drained from the peripheral tissues; the latter population of dendritic cells may function mainly for the purpose of maintaining tolerance4. However, when these immature lymph-node dendritic cells were exposed to antigen together with a maturation stimulus, they were able to activate T cells, suggesting that lymph-node dendritic cells could be potential targets for immunomodulatory agents4 (approaches for targeting the lymph node are described below). Also, it has been shown that dendritic-cell presentation of peptide-antigen-loaded MHC class II molecules in the draining lymph node of a tissue following subcutaneous antigen delivery came in two discrete stages: first, by the lymph-node dendritic cells, which acquired antigen in the lymph node; and, second, by the dendritic cells that had migrated there from the injection site10.
Further in support of the lymph node as a target, the humoral response is apparently initiated by lymph-node B cells, which take up antigen directly in the lymph-node follicles, rather than by migrating B cells or by dendritic cells, indicating that lymph-node targeting may be advantageous for protective antibody-generating vaccines11. This approach has not been widely investigated, and it remains to be determined how the T-cell response differs when initiated by peripherally activated dendritic cells and when initiated by lymph-node dendritic cells, as well as how the chemokine balance shifts in the lymph node and how this ultimately affects the long-term response. This is particularly important in light of emerging evidence demonstrating the importance of lymph-node T-cell homing in tolerogenesis12,13. Thus, although lymph-node targeting for immunomodulation has interesting prospects, much more research is needed.
The mucosal epithelia are a major site of immunosurveillance, responding to most immune challenges encountered by an organism. The mucosae of the airways, the digestive tract and the vagina, among others, contain special lymphoid tissues, referred to as the mucosa-associated lymphoid tissues. In these tissues, antigens are collected by microfold (M) cells and transferred to dendritic cells, which in collaboration with T cells induce a specialized humoral response of secretory IgA in the mucosal secretions, as well as a cellular response14. Much of the protective response of the mucosa derives from secretory IgA. Systemic vaccination, for example by intramuscular injection, does not typically induce strong mucosal immunity, whereas vaccination of the mucosa does, although in a region-specific manner14. Given the importance of inducing mucosal immunity and protection against the vast number of pathogens that enter through these routes, the different mucosae are important tissue targets, and the M cells and dendritic cells in the mucosa-associated lymphoid tissues are important cellular targets14.
In addition to understanding the tissue and cellular targets for immunotherapeutics, it is important to understand the subcellular targets. APCs process antigen from the cytosol or following endocytosis for display in MHC class I molecules and/or MHC class II molecules, respectively15. Intracellular proteins (for example those produced by viral pathogens) are degraded by the proteasome, released into the cytosol and subsequently translocated into the endoplasmic reticulum by the transporter associated with antigen processing (TAP) to be loaded into the peptide-binding site of MHC class I molecules1. By contrast, proteins internalized from the extracellular environment (for example from phagocytosed bacteria) are digested in endolysosomes, and peptides derived from these are loaded onto MHC class II molecules, which are then transported to the plasma membrane1. Some subsets of dendritic cells, classically CD8+ dendritic cells, can also efficiently present exogenously obtained peptides on MHC class I molecules, a process known as cross-presentation, making dendritic cells unique among the APCs (macrophages can also cross-present antigen but with much lower efficiency1). As mentioned above, CD8+ T cells are primed by interactions between the T-cell antigen receptors (TCRs) of CD8+ T cells and MHC class I molecules, and CD4+ T cells are primed by the binding of their TCRs to MHC class II molecules. Therefore, the choice of the route of antigen delivery, and thus which class of MHC molecule presents the antigen, directly (but not solely, as the nature of dendritic-cell activation by danger signals is also important) controls the type of antigen-specific T-cell response that is obtained. These presentation pathways, in their simplest and most classic form, are illustrated in Fig. 2.

A simplified view of antigen presentation by dendritic cells. Left, exogenous particles, proteins or pathogens can be taken into the cell through various pathways, including phagocytosis (for particles >1 μm), macropinocytosis (<1 μm), and endocytosis from caveolae (∼60 nm) or clathrin-coated pits (∼120 nm). Exogenous antigens are then processed in endocytic vesicles (phagosomes, endosomes, lysosomes and/or endolysosomes; dashed arrows represent multiple vesicular steps). Processed antigen (peptide) is subsequently loaded onto MHC class II molecules (which have been assembled in the endoplasmic reticulum, transported through the Golgi apparatus and targeted to endocytic compartments) in a lysosome or MHC class II compartment (MIIC). The peptide–MHC class II complexes then move through exocytic vesicles to the cell surface, where antigen presentation occurs. MHC class II loading of endogenous antigen provided by autophagy can also occur, particularly when the cell is under stress. Right, antigen can be loaded onto MHC class I molecules through two main pathways. In the classical pathway, endogenous or viral proteins in the cytosol are processed through the proteasome, transported into the endoplasmic reticulum through the molecule TAP (transporter associated with antigen processing), loaded onto MHC class I molecules, and then transported through the Golgi apparatus and exocytic vesicles to the cell surface for presentation. In addition, exogenous antigens that have been phagocytosed, macropinocytosed or endocytosed can be cross-presented on MHC class I molecules by some subsets of dendritic cell. In this pathway, antigen either may be loaded in endocytic compartments (not shown) or may escape endosomes and arrive in the cytosol, where it is processed through the proteasome as usual, loaded onto MHC class I molecules and transported to the surface. Finally, terminal degradation pathways can occur (for example when apoptotic cells are internalized). See refs 1 and 96, 97, 98 for details about antigen processing.
Materials as tools to modulate immune-cell function
As mentioned, APCs — in particular dendritic cells — are responsible for integrating a myriad of external biomolecular stimuli to produce an adaptive immune response. Antigen presentation leads to immunogenicity only when costimulatory molecules are presented with the MHC-class-I-associated or MHC-class-II-associated peptide antigen. Thus, to interpret what to do with the antigen, dendritic cells use receptor systems, such as pattern-recognition receptors (PRRs), that can sense either pathogen-derived or endogenous danger signals16,17. A pathogen may be recognized by several PRRs either simultaneously or sequentially, activating distinct or shared signalling pathways. How the dendritic cell responds, and therefore the quality of the induced adaptive immunity, is determined by the danger signals to which the dendritic cell is exposed17.
PRRs largely recognize pathogen-derived biomolecules referred to as pathogen-associated molecular patterns (PAMPs), which are evolutionarily distant non-self molecules such as LPS and viral double-stranded RNA18. Many endogenous molecules can also trigger PRR activation; such molecules, known as danger-associated molecular patterns (DAMPs), are typically associated with tissue damage or distress. A select group of ligands for a few representative PRRs, along with the immunological responses they induce, are described in Table 2.
Recognition of PAMPs and DAMPs by PRRs occurs both through extracellular activation cascades such as the complement system and through intracellular signalling pathways that can be initiated at the dendritic-cell surface, in the endosome after phagocytosis or in the cytosol16. PRR expression patterns vary significantly between subcellular compartments, immune-cell types and subtypes, and tissues16,19. One of the major PRR classes is the Toll-like receptor (TLR) family, members of which recognize a large number of pathogen-derived ligands and a smaller number of endogenously derived ligands. Of the TLRs, some — such as TLR4 and TLR2, which recognize LPS and lipoteichoic acid respectively — may be expressed on the plasma membrane. By contrast, others — such as TLR7, TLR8 and TLR9, which recognize bacterial RNA or DNA16 — may be present in the endosomal compartment. Many other intracellular and membrane-expressed PRRs are involved in the recognition of viral nucleic acids or bacterial and fungal carbohydrates by dendritic cells; these include cytosolic NOD-like receptors (such as NALP3), which activate the dendritic-cell inflammasome in response to bacterial and endogenous danger signals16. Again, the dendritic cell integrates these signals to determine whether to mature, how to process and present the antigen, and which cytokines to produce.
For these reasons, one important task in immunobioengineering is to develop delivery strategies by which to present antigen — along with PAMPs to ligate particular PRRs — so as to induce a desirable TH1-type or TH2-type adaptive immune response. We contemplate the targeting and penetration of barriers as objectives for materials design: the barrier of the antigen being able to find, or being found by, the APCs that reside in the tissues performing surveillance for signals of infection, the barrier of the tissue interstitium after injection of antigen into connective tissue such as skin, the barrier of the mucosa after antigen is sprayed into the nasal sinus or inhaled into the lungs, the barrier of entry to the endolysosomal compartment of the cell after endocytosis, and the barrier of entry to the cytosol presented by the endosomal membrane. Materials with different design principles and characteristics are being developed to accomplish these delivery tasks.
Materials for enhancing antigen uptake by APCs
Materials design considerations for enhancing uptake by APCs have focused on recognition and recruitment. With regard to recognition, some subclasses of dendritic cell possess an endocytic receptor, DEC205 (ref. 20), which has been successfully used to enhance dendritic-cell uptake, for example with an antigen or a biomaterial particle conjugated to anti-DEC205 antibodies21,22. Recruitment involves chemoattracting other APCs to the delivery site, and strategies include the use, variously, of degradable polyester particles to create gradients of dendritic-cell chemoattractants23, injectable hydrogels24, and degradable scaffolds that simultaneously release cytokines, PAMPs and antigen25. As an extreme example, cell transplantation approaches involve isolating dendritic cells from a subject, exposing them to antigen in vitro (referred to as 'loading'), stimulating them and then re-implanting them26.
Materials for penetrating tissue barriers
Whereas the section above describes approaches to delivering the dendritic cell to a material by targeting, recruitment or transplantation, approaches are also being explored to take the material directly to the endogenous dendritic cells in the draining lymph node after injection into the tissue interstitium, with particle size being the primary material control parameter. In most tissues, there exists a slow interstitial flow from the blood capillaries to the lymphatic capillaries, of the order of 0.1–1 μm s−1 (ref. 27). It is through this interstitial flow that macromolecules are swept into the lymphatics, and that immature lymph-node dendritic cells (and follicular B cells, as described earlier11) can sample self molecules and pathogen-derived molecules present in the tissues. This approach to targeting APCs is highly robust, as long as the immunotherapeutic particles are neither too big (resulting in entrapment in the interstitium) nor too small (resulting in absorption into the blood). The dependence of targeting on size has been probed with biomaterial nanoparticles sterically stabilized with polyethylene glycol (PEG) brushes: particles of ∼20–25 nm in diameter were very efficiently delivered to lymph nodes; particles of 45–50 nm in diameter were somewhat less efficiently delivered (64%, plus the 20–25-nm nanoparticles); and particles of 100 nm in diameter were poorly delivered (8%, plus the smaller particles)28. It is interesting to compare these sizes with those of viruses: the smallest viruses (for example the single-stranded DNA parvovirus or the RNA picornaviruses such as poliovirus) are in the 20–30-nm range, but most are substantially larger (for example adenoviruses, 70–80 nm; retroviruses, 100–200 nm; and poxvirus, 100 × 200 × 300 nm3)29. As described below, it is possible to use materials chemistry to access sizes as low as, and even smaller than, those of biological viruses.
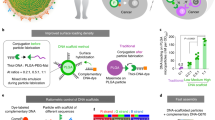
Three general material-fabrication schemes are useful for forming polymer nanoparticles in the 20–30-nm range: emulsion polymerization, self-assembly and branched-polymer synthesis (other techniques are available for inorganic nanoparticles). These schemes are illustrated in Fig. 3. In emulsion polymerization30, surfactant micelles are formed in an aqueous environment, and hydrophobic monomer is loaded within the micelle and polymerized. The result is a surfactant-stabilized polymer nanoparticle, the size distribution of which can be controlled by the ratio of surfactant to monomer. The particles contain a hydrophobic core within a hydrophilic corona (from the surfactant), to the surface of which hydrophilic molecules such as antigens and PAMPs may be conjugated31. Useful surfactants include block co-polymers, such as PEG-bl-polypropylene glycol-bl-PEG, also known as Pluronics28.

a, Emulsion polymerization is carried out in a continuous phase, with an emulsifier, here shown as an ABA block co-polymer (A in purple, and B in green), surrounding monomer droplets (centre). The surfactant PEG-bl-polypropylene glycol-bl-PEG is a convenient emulsifier, in that the terminal hydroxyls on the polymer may be used for antigen grafting (not shown), for example as in ref. 28. b, Self-assembly of amphiphilic block co-polymers can yield very small nanoparticles (as depicted). Diameters of less than 15 nm have been achieved33 using PEG-bl-polypropylene sulphide (PEG-bl-PPS). c, Even smaller structures for antigen display can be formed by synthesizing branched polymers, such as the dendrimer depicted. For further examples of dendrimers, see ref. 37. In all panels, antigen and danger signals may be attached to the nanoparticle surfaces. Note that the structure in c is much smaller than those in a and b.
Self-assembly is a powerful method for forming very small particles. In principle, the size distribution of particles formed by self-assembly can be very narrow, owing to the potential to approach equilibrium. Typically, amphiphilic block co-polymers are dissolved in a water-miscible organic medium that is a solvent for both block compositions, and this solution is subsequently dropped into water, which is a solvent for one block but not the other, forcing micellization with the hydrophobic block at the core of the micelle. Polymer micelles are intrinsically unstable structures that can disassemble after the infinite dilution that follows injection into the body, driving considerations of how to engineer an optimal dissociation rate.
The hydrophobicity of the core-forming block is an important consideration; for example, the critical micelle concentration of block co-polymers containing polypropylene glycol (such as Pluronics) is not as favourable (low) as that of analogous block co-polymers containing polypropylene sulphide (PPS), in which the oxygen atoms in the polymer backbone of polypropylene glycol have been replaced with sulphur atoms32. Using this materials chemistry, it is possible to access the subviral size range; for example, PEG44-bl-PPS10 forms spherical micelles that have 7-nm cores and 14-nm total diameters33 and that demonstrate slow dissociation. Alternatively, very high-molecular-weight polypropylene glycol (the hydrophobic block) in Pluronics micelles can serve as a stabilizing influence34.
In addition to hydrophobicity, and the molecular size of the hydrophobe in a self-assembling block co-polymer, the melting temperature (Tm) and the glass-transition temperature (Tg) are important. On the one hand, low-Tg polymers have the advantage of being readily formed at normal production temperatures, thus approaching equilibrium micelle size and shape; on the other hand, higher-Tg hydrophobic block compositions (Tg >37 °C) or crystalline hydrophobic polymers (Tm >37 °C) self-assemble at supraphysiological temperatures for very stable use at 37 °C, below Tg or Tm. Micelle, and also polymersome35 (see below), processability can thus be engineered by manipulating any of the material parameters mentioned above to exploit the equilibrium nature of the materials, as well as by slowing the dissociation rate to suit practical use.
Biologically derived molecules may also readily self-assemble, as can be observed in virus-like particles, which are now in clinical use. Virus-like particles are biotechnologically produced, self-assembled structures of viral capsid proteins and can approach the 50-nm diameter range36. Heterologous production of the viral capsid proteins ensures their self-assembly without a viral genome, and self-assembled protein particles therefore possess the great advantage of biological functionality and intrinsic immunoreactivity, without the potential for infectivity. As these materials are biologically derived, however, they are more expensive and can be stored less stably than synthetic materials. Nevertheless, they are clinically highly effective and serve as an excellent model to be mimicked by biomaterials scientists.
Branched polymers, especially dendrimers, have been developed as very small nanoparticles to display antigen and danger signals37. The advantages of branched-polymer strategies include the stability ensured by the covalent bonds within the polymer and a very narrow size distribution due to the nature of their synthesis. This synthesis is performed stepwise from a multifunctional initial scaffold, by adding difunctional monomer in serial couple–deprotect steps. Thus, the number of end groups on the dendrimer doubles with each serial step. As well-characterized display systems of well-defined and controllable size, these materials have great potential as vaccine platforms38.
Other materials design considerations for polymer particles intended for eventual therapeutic use relate to materials stability and elimination. Although a degradation mechanism is necessary, ensuring stability during storage is paramount. It is very difficult to dry and then resuspend nanoparticles at their original size distribution; moreover, gentle drying processes such as lyophilization are expensive, which may hinder the use of a vaccine form in global applications. Therefore, storage in water, preferably without refrigeration, is an important design goal. Promising materials chemistries have been explored to engineer nanoparticles that degrade either by oxidation or reduction (requiring that the nanoparticles be stored in a controlled atmosphere, which is inexpensive) or by pH-triggered hydrolysis (requiring storage at a stable pH). Also, simple dissociation to form unimolecular final products of a molecular weight low enough for renal clearance (less than ∼10,000 g mol−1) is a feasible approach (requiring storage above the critical micelle concentration)39. These and other chemistries will be introduced in more detail below.
Materials for penetrating mucosal barriers
Mucosal surfaces present a target for vaccination that is both appealing and challenging. Most pathogens invade the body through a mucosal route, be it the nasal cavity, the airways, the gut, the vagina or the rectum; therefore, establishment of a secretory IgA immune response in these tissues would be especially beneficial for providing protection. Delivery of vaccines to these surfaces is substantially complicated by the mucosal layer that otherwise protects them, at least in part, against pathogen entry. The mucus consists of a physically crosslinked, viscoelastic hydrogel, with mesh sizes of the order of 10–100 nm (ref. 40). Barrier penetration is largely restricted for particles that are greater in diameter than a few hundred nanometres40,41, although particles that are ∼50 nm in diameter can diffuse in mucus almost as freely as they do in water40.
Particle surface properties have a major role in particle penetration of mucus. It has been observed that even very large particles, 200–500 nm in diameter, can penetrate mucus when appropriately grafted with surface PEG chains42. This effect depends strongly on the molecular weight of the grafted PEG, with grafts comprising shorter chains (2,000 g mol−1) penetrating well but those comprising longer chains (10,000 g mol−1) penetrating several orders of magnitude more slowly. The field of mucoadhesion has been explored from a polymer science perspective: surface-tethered polymer chains interpenetrate, and entangle within, the mucin polymer network, leading to adhesion associated with entanglement and disentanglement43, and longer chains interpenetrate more effectively than shorter ones44. These concepts are illustrated in Fig. 4. Here, mucoadhesion is not beneficial, as the vaccine particles become entrapped in the mucosal barrier; therefore, PEG chains long enough to prevent adsorption, but not long enough to lead to entanglement, are desired.

Nanoparticles need to gain access to the mucosal epithelia for antigen delivery or transfection, so they must be able to penetrate the mucous layer. The grafting of polymers such as PEG (pink) to nanoparticles (yellow) has been explored as a way of blocking the adsorption of particles to components of the mucus (green). Studies into mucosal bioadhesion have examined various physical regimes of polymers. Shorter, denser graft layers tend to sterically stabilize the nanoparticle surface (a). By contrast, longer, sparser grafts allow interpenetration of the two polymers (the grafted chains and the mucous network) (b), leading to adhesion to the mucus43,44,99 and unfavourable nanoparticle penetration42,100.
Gene vectors, which are useful in DNA vaccination (that is, with the antigen being expressed from the delivered DNA), present a particular challenge in mucosal penetration, in that most nanoparticle complexes containing DNA are formed with cationic carriers, such as cationic lipids; these cationic charges can dramatically limit particle transit through the negatively charged mucous layer45. Cationic lipid mixtures have been developed with PEG-grafted lipid components to enhance particle penetration46, although the polymer chains may interfere with later processes of endosomal destabilization and gene uptake. To address this, vectors with PEG chains that are removed by cellular processes (see the next section) during endosomal processing are being investigated47.
Materials for intracellular targeting
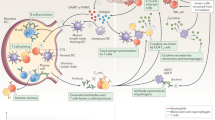
As described above, the detection of both antigens and danger signals is complex and takes place in different compartments of the cell. For example, antigens for presentation by MHC class I pathways must be available within the cytosol, whereas those for presentation by MHC class II molecules must be present within the endolysosomal compartment (Fig. 2). With regard to danger signals for APC activation, even considering just the TLR family, receptors for some ligands (such as hydrophobic bacterial cell-wall components, which bind to TLR4, or bacterial flagellae components, which bind to TLR5) are present on the plasma membrane, whereas receptors for others (single-stranded RNA, which binds to TLR7, or unmethylated bacterial CpG DNA, which binds to TLR9) are present and active within the endolysosome. The spatial details of antigen and danger-signal delivery are therefore important. Several polymers that accomplish endolysosomal delivery are described below and are shown in Fig. 5.

a–d, Polymers sensitive to oxidation within the lysosome (a) or reduction within the endosome (b–d). a, Oxidation of a hydrophobic sulphide, ultimately to a hydrophilic sulphone, leads to dissociation of self-assembled vesicles of the macroamphiphiles, which after oxidation are only hydrophilic and no longer amphiphilic48. b, Reduction of a disulphide link between the hydrophobic and hydrophilic blocks of a vesicle-forming macroamphiphile leads to vesicle rupture35. c, Reduction of an AB multiblock polymer leads to dissociation of a complex with DNA in the endosome50. d, Reduction of an analogous peptide-crosslinked DNA particle leads to DNA release51. e–f, Polymers sensitive to hydrolysis during acidification of the endolysosome, utilizing orthoesters (e) and ketals (f). R and R′ are usually alkyl groups, to adjust the hydrophobicity of the material.
A number of chemical reactions have been engineered to release particle payload within the endolysosomal compartments. As the antigen in the endosomal vesicles is processed and the vesicles mature towards lysosomal fusion, the intravesicular pH is lowered, first to a pH of ∼6 in the endosome and then to a pH of ∼5 in the lysosome, relative to the extracellular pH, 7.4. Additionally, the reduction–oxidation state of these compartments changes: the endosome is rendered reductive, whereas the lysosomal compartment is substantially oxidative, compared with the mildly oxidative extracellular environment. For these reasons, both pH-sensitive and reduction–oxidation-sensitive materials are being studied.
Oxidation at low pH represents the final stages of endolysosomal processing, with exposure in the lysosome to a number of reactive oxygen species. Oxidation-sensitive dissociation of self-assembling block co-polymers has been engineered, by designing block co-polymers (mentioned above) containing a hydrophobic PPS block; on exposure to oxidative conditions, the block is converted to hydrophilic polypropylene sulphoxide and, ultimately, to the more hydrophilic polypropylene sulphone48. When block co-polymer architectures are selected with approximately equal block volumes, so as to form vesicles known as polymersomes, on oxidation the polymersomes transform into worm-like micelles, then into spherical micelles and finally into soluble polymer, as the ratio of the effective volumes of the hydrophobic block and the hydrophilic regions decreases eventually to zero49, releasing the contents of the polymersome.
Earlier in the processes of endolysosomal processing, endocytosed nanoparticles encounter a reductive environment. Self-assembling block co-polymers with architecture PEG-SS-PPS, that is with a reducible disulphide connection between the hydrophobic and hydrophilic blocks, have been shown to destabilize within 15 min of endocytosis in a macrophage-like cell line (a model of APCs), releasing the contents of the polymersomes within the early endosome35. Linear multiblock co-polymers have also been developed; they consist of DNA-binding peptides that are flanked on each side by a cysteine residue and polymerized by oxidation in vitro50. The resultant linear, high-molecular-weight polymer binds DNA and condenses it into a nanoparticle, referred to as a polyplex, but reduction within the endosome leads to multiple chain scission to form products that bind only weakly to DNA, thereby releasing the material. If endosomal disruption is also accomplished (see below), efficient transfection with antigen-encoding DNA can be achieved50. Moreover, if nucleotide-based PAMPs, such as CpG oligonucleotides, are included, efficient delivery to the endosomal receptors can be achieved. Low-molecular-weight disulphide peptides have also been used to good effect as reduction-sensitive crosslinkers for the endosomal release of polyplexes51.
The pH gradient experienced during endolysosomal processing has also been used for endosomal release. Whereas simple degradable polyesters such as polylactic acid, polyglycolic acid and their co-polymers are degraded by acid-catalysed hydrolysis, the hydrolysis rates at even lysosomal pH are so slow that they are not very useful for endosomal delivery. For this reason, more acid-sensitive, hydrolytically sensitive links, such as orthoesters52 and ketals53, have been sought. For example, particles crosslinked with ketal moieties have been developed for biomolecular delivery53, including vaccination54, through the targeting of endosomal release. Endosomal release of CpG oligonucleotides has also been shown to be feasible using these materials chemistries55. Degradation rates at lysosomal pH can be ∼20-fold faster than at extracellular pH55, providing the desired trigger.
Whereas the endosomal compartment is the interesting target for MHC class II loading, MHC class I presentation requires the antigen payload to be present in the cytosol. Thus, disruption of the endosomal membrane barrier to access the cytosol is an important target. Endosomal disruption is also necessary for DNA vaccination, in which plasmid DNA must be expressed to produce the antigen. Materials schemes that have been investigated for these purposes are illustrated in Fig. 6.

a, Polypropyl acrylic acid disrupts membranes in a pH-dependent manner, with membranolytic activity at about the pH of early endosomes (∼6.5). A block co-polymer is shown, of polypropyl acrylic acid and a monomer linked through a disulphide bond to the antigen, which can be released in the reductive environment of the endosome56. b, Crosslinked particles that are hydrolytically sensitive at endosomal pH, releasing a cell-penetrating peptide (CPP, which consists of polyarginine57), are formed by inverse emulsion polymerization of acrylamide, a CPP-grafted acrylamide and a ketal-containing bisacrylamide crosslinker, yielding the polymer shown here. Hydrolysis both degrades the crosslinks in the polymer and releases the CPP, destabilizing the endosome. c, The proton-sponge effect58 for endosomal disruption, and thus cytosolic delivery, has been implemented in core–shell nanoparticles by first polymerizing particles of diethylaminoethyl methacrylate and then sequentially polymerizing aminoethyl methacrylate, both with crosslinking59. One monomer forms the core (left), and one forms the corona, or shell (right).
Both reductive and pH triggers have been used to control endosomal escape. With regard to reduction, the polymer fragments produced by reduction of the PEG-SS-PPS block co-polymers mentioned above apparently possessed sufficient surface activity to disrupt the endosomal membrane, making the payload available within the endosome within 15 min but within the cytosol within 2 h (ref. 35). Triggers sensitive to pH are more commonly sought. One approach involves the engineering of polymers that express pH-dependent surface activity and thus membrane-disruptive activity, for example polypropyl acrylic acid; these polymers display no membrane-disruptive activity at extracellular pH, but at pH 6–6.5 are strongly membranolytic56. These polymers have demonstrated strong potential for antigen delivery, processing, and presentation by MHC class I molecules56. Oligocations and polycations can also destabilize the cell membrane; cationic cell-penetrating peptides based on oligoarginine have been incorporated in pH-sensitive ketal-crosslinked nanoparticles — the membranolytic moieties becoming available as the pH is lowered and hydrolysis proceeds — with beneficial effect for intracellular accumulation57.
Polycations can have an additional favourable effect on membrane destabilization, owing to an osmotic imbalance that ensues during their protonation. Polyethylene imine possesses a favourable pKa value for protonation within the endosome, the associated osmotic effect being referred to as the 'proton-sponge' effect58. The use of such polymers is, however, hampered by cytotoxicity associated with polyethylene imine's contact with the cell's membranes. To retain the proton-sponge effect yet eliminate cytotoxicity, core–shell nanoparticles have been studied using sequential emulsion polymerization, first of a secondary-amine-containing monomer of appropriate pKa (diethylaminoethyl methacrylate), to form a core, and then of a second monomer, to form a corona (or shell)59. Regional separation of the functions of the particle led to favourable cytosolic release, through endosomal disruption by means of the proton-sponge effect, with markedly lower cytotoxicity than caused by free polyethylene imine60.
Materials interaction with, and penetration of, cell membranes to access the cytosol is complex, involving hydrophobic as well as electrostatic interactions. One new class of particle has demonstrated intriguing potential, based on an intrinsic membrane-penetrating ability derived from its spatial distributions of hydrophobicity and charge. Each particle consists of a spatially heterogeneous organic adsorbed layer formed atop a 6-nm gold nanoparticle to create <1-nm striations of hydrophobicity and negative charge61. These particles, but not those with the same overall charge density but lacking the striations, were able to pass directly through the plasma membrane. This concept, if it can be used as a general design principle for other particle implementations, will be very powerful.
The cytosol represents a particularly attractive target for antigen-encoding DNA, as well as for protein antigen. If materials approaches to cytosolic targeting can allow highly efficient DNA delivery concurrently with prolonged PRR activation, dendritic cells could become a very attractive target for DNA vaccination. Given the logistical advantages of delivery of antigen-encoding DNA (speed of production, cost and stability) relative to those of delivery of protein antigen, the attraction of such approaches for counteracting pathogens that vary seasonally (such as influenza virus) and pathogens that mainly affect the developing world is considerable.
Materials to trigger immune-specific functions
Now that we have introduced the systems by which to deliver antigens and danger signals to specific cellular and subcellular compartments, we consider how the materials affect dendritic cells either directly or indirectly, for example through mediation by protein–material interactions or through a delivered biomolecular payload (although much of this, with the exception of the antigen, has been addressed above). Because PRRs are fundamental to the initiation of immunity, owing to their mediation of the recognition of PAMPs and DAMPs, biomaterials are being implemented to deliver natural or synthetic immunomodulatory agents to these receptors. However, the material itself may also be intrinsically biologically active, by virtue of its particulate character or as a result of protein interactions at the biomaterial surface.
The role of particle size and shape
As discussed above, particle size can control the biological transport and hence the bioavailability of a material to a remarkable degree, for example through the tissue interstitium or across a mucosal barrier. However, material size characteristics may also be an important determinant of a material's immunological activity. For example, the most common adjuvants in clinical use are insoluble aluminium phosphates (alum), which form aggregates with the protein antigen and create an antigen depot. These particles, which might be considered chemically inert, enhance cellular IL-1β secretion62 by means of NALP3 (NACHT domain-, leucine-rich repeat-, and PYD-containing protein 3, also known as NLRP3; ref. 63) to induce antibody-mediated protective immunity; therefore, the particles are themselves intrinsically recognized as a sign of danger. Because material particles can be both effective and economical, understanding and engineering this effect is of great interest. Recent work suggests that this is possible, with evidence that polymeric microparticles endocytosed by dendritic cells trigger the inflammasome by means of NALP3 and, in concert with endogenous signals, induce both humoral and cellular immunity64. In vitro, inflammasome-activation-associated IL-1β secretion by dendritic cells in response to particle treatment (in addition to LPS stimulation) was size dependent and maximal at particle diameters between 400 and 1,000 nm (ref. 64). Given the <100-nm size limit for interstitial transport to access the lymph nodes, where large number of dendritic cells reside, the engineering of nanoscale particles to activate the inflammasome efficiently warrants further study.
Particle shape may also be important; for example, for micrometre-scale particles the ability of macrophages to phagocytose a particle depends more on its shape than on its size65. This phenomenon is determined by actin mechanics at the points of particle contact. These results raise the intriguing possibility that shape may have such a role, either in uptake or in APC activation, at shorter length scales as well.
The role of particle hydrophobicity
Within the diverse family of TLR4-ligand and TLR2-ligand PAMPs (including LPS, lipopeptide and peptidoglycan) and DAMPs (including hyaluronan fragments, heat-shock proteins and fibronectin), an underlying biochemical thread may be the presence of hydrophobic domains, suggesting hydrophobicity to be a universal sign of danger sensed by TLRs18. Exploiting this connection may be of great interest as a potentially economical strategy by which to ligate hydrophobicity-sensing PRRs and possibly avoid costly conjugation schemes for recombinant protein ligands or synthetic PRR ligands. The supramolecular organization of biomacromolecules that contain hydrophobic domains and have the capacity to hide or expose these hydrophobic patches seems to determine their ability to activate immunity18.
Given that microparticle formulations can be formed by nanoscale self-assembly of hydrophobic microdomains, approaches could be designed to control the degree of nanoparticulate hydrophobic-domain exposure, to exploit such a mechanism. However, the task of preserving a hydrophobic material surface once exposed is difficult: in the presence of protein in the biological environment, protein adsorption can rapidly obscure hydrophobic interfaces. From the perspective of inducing immunity, adsorption may have some advantages; the roles of immunoregulatory molecules (such as scavenger receptors and complement component C1q) in controlling normal hydrophobic molecule transport and clearance rather than in initiating an immune response18, as well as in molecular recognition specificity in TLR4 ligation rather than in TLR2 ligation (as these ligands induce TH1-type and TH2-type responses, respectively), remain to be elucidated to determine materials design guidelines for such a strategy. Thus, materials hydrophobicity may itself be a danger signal or may be interpreted by dendritic cells through the intermediating layer of adsorbed plasma proteins.
Complement activation
Whereas hydrophobicity in materials modulates relatively nonspecific protein adsorptive interactions, certain materials features can mimic features of pathogen surfaces to activate innate immune pathways. One such recognition cascade is complement, the alternative pathway of which recognizes certain primary hydroxyls66 and other surface nucleophiles67 to react at the site of a strained thioester in C3, forming material-bound activation product C3b68. Although much biomaterials research seeks to avoid such interactions, immunobioengineering can exploit complement activation, particularly in light of the variety of ways in which complement can affect innate and adaptive immunity69,70. Notably, the C3 activation products C3d and C3b have been shown to be molecular adjuvants capable of inducing strong antigen-specific humoral immunity71,72,73, and materials have been developed to exploit C3 activation for adaptive immunity28.
Studies suggest that surface biochemistry such as sulphation may control the activation and deposition of complement species on cellular or material surfaces by controlling the adsorption of complement factor H (CFH) and CFD74,75. Generating complement-opsonized particulates in situ by modulating material surface chemistry may therefore represent an inexpensive and powerful strategy to harness the molecular adjuvant properties of C3b and C3d. Interestingly, C1q, which binds to immunoglobulin on antigen ligation, also binds to hydrophobic molecules or aggregates such as LPS and liposomes18. Hence, the incorporation of hydrophobic domains could activate complement through the classical pathway to use the immunomodulatory properties of C2 and C4.
Functionalization and encapsulation
We have seen that the biological context in which the targeted PRR is encountered by the material (for example at the cell surface or in the endosome) must be considered when designing materials delivering bioavailable molecules for PRR ligation. In addition, the benefits of antigen display relative to encapsulation and of danger-signal co-encapsulation relative to co-delivery must be considered. For polymers that degrade too slowly for antigen encapsulation schemes, such as polylactic-co-glycolic acid, adsorption of antigen onto biomaterial particles may be more beneficial76. Within these systems, co-encapsulation of PAMPs, such as CpG oligonucleotides or TLR4 ligands, is much more beneficial than co-administration, providing support for prolonged stimulation of dendritic cells after antigen collection77,78,79,80. Indeed, when dendritic cells encounter both self antigen and pathogen-derived antigen, they use the coexistence of antigen and TLR ligand within the same endosome to distinguish between the two and enhance presentation of the pathogen-derived antigen on MHC class II molecules81. Bringing the antigen together with the PAMP, so as to model the situation found in the pathogen, therefore seems to be a sound design principle.
Materials as models for basic immunobiology
When immune cells interact with pathogens, or with each other, they do so in a complex display of a number of molecular mediators of antigen uptake, processing, presentation and activation. The challenge of teasing out molecular mechanisms from this process in its full complexity can be daunting. Materials science in immunobioengineering allows the development of tools with which to assess molecular and cellular hypotheses. Whereas the sections above highlight research goals with translational ends, here we briefly touch on some that are more basic and mechanistic.
One opportunity for biomaterials research in immunobioengineering is the creation of synthetic pathogens for the study of dendritic-cell activation and downstream interactions with T cells that trigger adaptive immunity. Because dendritic cells have evolved to recognize such a diverse array of PAMPs, and because a number of such PAMPs are found in any one pathogen, it is difficult to study the molecular interactions in simple, isolated systems. Biomaterials can provide a blank slate on which antigen and a defined set of PAMPs can be displayed in defined amounts for mechanistic investigation. As an example, we refer to the previously mentioned study on nanoparticle-induced activation of complement by means of surface-tethered PEG, in which complement was the only DAMP in the absence of any pathogen-associated signals28. Investigation of the pairwise interactions of particle size and complement activation demonstrated that complement is a powerful trigger of humoral and cellular immunity as long as the complement-decorated particles are small enough (<50 nm in diameter) to enter the lymphatics and thus target lymph-node APCs28. Many other such mechanistic studies could use biomaterials as model systems, controlling the destination of PAMP presentation (plasma membrane or endolysosome, or both) and the identity of the PAMPs (for example with multiple TLR ligands), with independent control of the numbers of PAMP molecules, their clustering and spatial organization and even their duration of exposure.
Investigating the interactions between immune cells themselves presents another interesting challenge for materials science in immunobioengineering. For example, when activated dendritic cells present their antigen to T cells, a spatially organized structure referred to as the immunological synapse is created, in which the peptide-antigen-loaded MHC molecule on the dendritic cell is presented to the TCR on the T cell; this receptor pair is organized after cellular contact such that the peptide–MHC–TCR pair (one receptor on each of the two cells) is clustered and is surrounded by an adhesion-receptor pair consisting of intercellular adhesion molecule 1 (ICAM1) on the dendritic cell binding to lymphocyte function-associated antigen 1 (LFA1) on the T cell82. It was previously unknown whether this evolving geometric patterning was required for the function of the immunological synapse or was only associated with it. Using supported lipid membranes in which bound peptide–MHC and ICAM1 could freely move laterally, a functional immunological synapse could be formed, but when barriers were created to limit lateral reorganization of peptide–MHC and ICAM1, function was inhibited83; this suggested that the biological structuring is required for function. Likewise, when a TCR ligand was lithographically patterned in spots surrounded by ICAM1, a functional immunological synapse resulted, whereas when the geometry was reversed, no such function was possible84. In both of these examples, sophisticated materials science approaches allowed well-controlled models of immune-cell interactions to probe biological hypotheses.
Future prospects
Materials science has a great deal to offer the field of immunology: immunobioengineering of prophylactic and therapeutic vaccine platforms, and model systems with which to explore the molecular and cellular interactions between immune cells and pathogens (and between different classes of immune cell), are just a few examples of this. The application of materials science towards therapy and prophylaxis is instructed by immunobiology, and we have shown how materials science can in return be used to instruct immunobiology. The key feature that materials offer is that of design: design for encapsulation, design for immobilization or release of one or several biomolecular regulators of immune interactions, design for material functionalities such as release or membrane disruption in particular parts of the cell, and design for doing this in particular cellular and tissue targets. Some avenues may be more fruitful than others. Functional polymersomes are particularly attractive, in that both antigen and danger-signal payloads may be incorporated in the watery vesicle core, and other danger signals or targeting ligands may be attached to the polymersome surface or within the hydrophobic leaflet of the membrane. With materials chemistries leading to disruption and release within the endosome, and even destabilization of the endosomal membrane itself, a means of delivery for presentation by both MHC class II molecules and MHC class I molecules seems within reach. Still more interesting is the possibility of combining such means with physiological routes for delivery, for example by using ultrasmall self-assembled nanostructures to deliver such advanced materials directly to the lymphatics or to lymphoid tissue associated with the mucosae.
Clearly, even with the objective of vaccination described above, much work remains to be done, in that only very early implementations are clinically available or are in advanced stages of testing, rather than still being researched. As our understanding of immunobiology grows, so will the range of principles for the design of materials and material–biomolecular conjugates used in immunotherapeutics. Moreover, the design principles will be different in various contexts: for example, in therapeutic vaccines against cancer, complex materials and formulations may be contemplated, as cost does not present a major consideration. By contrast, vaccination in a global context places severe constraints on logistics (wet formulations and unrefrigerated storage), use (preferably needle-free administration routes and few doses) and cost (materials that can be easily manufactured and contain a minimal number of biological molecules). Although constraints are an annoyance from a design perspective, they can also present exciting intellectual challenges for the materials scientist and the immunobioengineer. The opportunity to combine possibilities for translation with those of using materials systems to learn more about the underpinning science, immunobiology, is also tremendously exciting.
References
Trombetta, E. S. & Mellman, I. Cell biology of antigen processing in vitro and in vivo . Annu. Rev. Immunol. 23, 975–1028 (2005).
Steinman, R. M. & Hemmi, H. Dendritic cells: translating innate to adaptive immunity. Curr. Top. Microbiol. Immunol. 311, 17–58 (2006).
Perrigoue, J. G. et al. MHC class II-dependent basophil–CD4+ T cell interactions promote TH2 cytokine-dependent immunity. Nature Immunol. 10, 697–705 (2009).
Wilson, N. S. et al. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood 102, 2187–2194 (2003).
Randolph, G. J., Angeli, V. & Swartz, M. A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nature Rev. Immunol. 5, 617–628 (2005).
Dudziak, D. et al. Differential antigen processing by dendritic cell subsets in vivo . Science 315, 107–111 (2007).
Singh, A. et al. Efficient modulation of T-cell response by dual-mode, single-carrier delivery of cytokine-targeted siRNA and DNA vaccine to antigen-presenting cells. Mol. Ther. 16, 2011–2021 (2008).
Cubas, R. et al. Virus-like particle (VLP) lymphatic trafficking and immune response generation after immunization by different routes. J. Immunother. 32, 118–128 (2009).
Lammermann, T. & Sixt, M. The microanatomy of T-cell responses. Immunol. Rev. 221, 26–43 (2008).
Itano, A. A. et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 19, 47–57 (2003).
Pape, K. A., Catron, D. M., Itano, A. A. & Jenkins, M. K. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26, 491–502 (2007).
Förster, R., Davalos-Misslitz, A. & Rot, A. CCR7 and its ligands: balancing immunity and tolerance. Nature Rev. Immunol. 8, 362–371 (2008).
Schneider, M. A., Meingassner, J. G., Lipp, M., Moore, H. D. & Rot, A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J. Exp. Med. 204, 735–745 (2007).
Holmgren, J. & Czerkinsky, C. Mucosal immunity and vaccines. Nature Med. 11, S45−S53 (2005).
Heath, W. R. et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol. Rev. 199, 9–26 (2004).
Lee, M. S. & Kim, Y. J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76, 447–480 (2007).
Macagno, A., Napolitani, G., Lanzavecchia, A. & Sallusto, F. Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol. 28, 227–233 (2007).
Seong, S. Y. & Matzinger, P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nature Rev. Immunol. 4, 469–478 (2004).
Zarember, K. A. & Godowski, P. J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168, 554–561 (2002).
Jiang, W. et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 375, 151–155 (1995).
Nchinda, G. et al. The efficacy of DNA vaccination is enhanced in mice by targeting the encoded protein to dendritic cells. J. Clin. Invest. 118, 1427–1436 (2008). This paper gives a demonstration of a DNA vaccine encoding an antigen targeted at dendritic cells, through expression of an antigen fusion protein with single-chain variable antibody fragments directed against DEC205.
Kwon, Y. J., James, E., Shastri, N. & Fréchet, J. M. In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles. Proc. Natl Acad. Sci. USA 102, 18264–18268 (2005). In this paper, ketal-crosslinked acid-sensitive nanoparticles are functionalized with anti-DEC205 antibodies, providing multiple levels of nanoparticle functionality.
Zhao, X., Jain, S., Larman, H. B., Gonzalez, S. & Irvine, D. J. Directed cell migration via chemoattractants released from degradable microspheres. Biomaterials 26, 5048–5063 (2005).
Hori, Y., Winans, A. M. & Irvine, D. J. Modular injectable matrices based on alginate solution/microsphere mixtures that gel in situ and co-deliver immunomodulatory factors. Acta Biomater. 5, 969–982 (2009).
Ali, O. A., Huebsch, N., Cao, L., Dranoff, G. & Mooney, D. J. Infection-mimicking materials to program dendritic cells in situ . Nature Mater. 8, 151–158 (2009).
Hori, Y., Winans, A. M., Huang, C. C., Horrigan, E. M. & Irvine, D. J. Injectable dendritic cell-carrying alginate gels for immunization and immunotherapy. Biomaterials 29, 3671–3682 (2008).
Swartz, M. A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 50, 3–20 (2001).
Reddy, S. T. et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nature Biotechnol. 25, 1159–1164 (2007).
Zubay, G. Biochemistry 1052–1053 (Addison-Wesley, 1983).
Rehor, A., Tirelli, N. & Hubbell, J. A. A new living emulsion polymerization mechanism: episulfide anionic polymerization. Macromolecules 35, 8688–8693 (2002).
Rehor, A., Tirelli, N. & Hubbell, J. A. Novel carriers based on polysulfide nanoparticles: production via living emulsion polymerization, characterization and preliminary carrier assessment. J. Control Release 87, 246–247 (2003).
Cerritelli, S., Velluto, D., Hubbell, J. A. & Fontana, A. Breakdown kinetics of aggregates from poly(ethylene glycol-bl-propylene sulfide) di- and triblock copolymers induced by a non-ionic surfactant. J. Polym. Sci. A 46, 2477–2487 (2008).
Velluto, D., Demurtas, D. & Hubbell, J. A. PEG-b-PPS diblock copolymer aggregates for hydrophobic drug solubilization and release: cyclosporin A as an example. Mol. Pharm. 5, 632–642 (2008).
Todd, C. W. et al. Development of an adjuvant-active nonionic block copolymer for use in oil-free subunit vaccines formulations. Vaccine 15, 564–570 (1997).
Cerritelli, S., Velluto, D. & Hubbell, J. A. PEG-SS-PPS: reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules 8, 1966–1972 (2007).
Roy, P. & Noad, R. Virus-like particles as a vaccine delivery system: myths and facts. Hum. Vaccin. 4, 5–12 (2008).
Gillies, E. R. & Frechet, J. M. J. Dendrimers and dendritic polymers in drug delivery. Drug Discov. Today 10, 35–43 (2005).
Sheng, K. C. et al. Delivery of antigen using a novel mannosylated dendrimer potentiates immunogenicity in vitro and in vivo . Eur. J. Immunol. 38, 424–436 (2008).
Yamaoka, T., Tabata, Y. & Ikada, Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J. Pharm. Sci. 83, 601–606 (1994).
Cone, R. A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 61, 75–85 (2009).
Lai, S. K., Wang, Y. Y. & Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 61, 158–171 (2009).
Lai, S. K. et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl Acad. Sci. USA 104, 1482–1487 (2007).
Peppas, N. A., Hansen, P. J. & Buri, P. A. A theory of molecular diffusion in the intestinal mucus. Int. J. Pharm. 20, 107–118 (1984).
Huang, Y. B., Szleifer, I. & Peppas, N. A. Gel–gel adhesion by tethered polymers. J. Chem. Phys. 114, 3809–3816 (2001).
Sanders, N., Rudolph, C., Braeckmans, K., De Smedt, S. C. & Demeester, J. Extracellular barriers in respiratory gene therapy. Adv. Drug Deliv. Rev. 61, 115–127 (2009).
Sanders, N. N., De Smedt, S. C., Cheng, S. H. & Demeester, J. Pegylated GL67 lipoplexes retain their gene transfection activity after exposure to components of CF mucus. Gene Ther. 9, 363–371 (2002).
Meyer, M. & Wagner, E. pH-responsive shielding of non-viral gene vectors. Expert Opin. Drug Deliv. 3, 563–571 (2006).
Napoli, A., Valentini, M., Tirelli, N., Muller, M. & Hubbell, J. A. Oxidation-responsive polymeric vesicles. Nature Mater. 3, 183–189 (2004).
Napoli, A., Bermudez, H. & Hubbell, J. A. Interfacial reactivity of block copolymers: understanding the amphiphile-to-hydrophile transition. Langmuir 21, 9149–9153 (2005).
Manickam, D. S. & Oupický, D. Multiblock reducible copolypeptides containing histidine-rich and nuclear localization sequences for gene delivery. Bioconjug. Chem. 17, 1395–1403 (2006).
McKenzie, D. L., Smiley, E., Kwok, K. Y. & Rice, K. G. Low molecular weight disulfide cross-linking peptides as nonviral gene delivery carriers. Bioconjug. Chem. 11, 901–909 (2000).
Wang, C. et al. Molecularly engineered poly(ortho ester) microspheres for enhanced delivery of DNA vaccines. Nature Mater. 3, 190–196 (2004).
Paramonov, S. E. et al. Fully acid-degradable biocompatible polyacetal microparticles for drug delivery. Bioconjug. Chem. 19, 911–919 (2008).
Cohen, J. A. et al. T-cell activation by antigen-loaded pH-sensitive hydrogel particles in vivo: the effect of particle size. Bioconjug. Chem. 20, 111–119 (2009).
Heffernan, M. J., Kasturi, S. P., Yang, S. C., Pulendran, B. & Murthy, N. The stimulation of CD8+ T cells by dendritic cells pulsed with polyketal microparticles containing ion-paired protein antigen and poly(inosinic acid)-poly(cytidylic acid). Biomaterials 30, 910–918 (2009). In this paper, acid-labile microparticles are developed to degrade quickly after endocytosis, releasing antigen and a ligand for an intracellular triglyceride-rich lipoprotein.
Flanary, S., Hoffman, A. S. & Stayton, P. S. Antigen delivery with poly(propylacrylic acid) conjugation enhances MHC-1 presentation and T-cell activation. Bioconjug. Chem. 20, 241–248 (2009). In this paper, a pH-sensitive polymer is demonstrated to destabilize the endosomal membrane after endocytosis, allowing access of an associated protein antigen to the cytosol and therefore MHC class I presentation.
Cohen, J. L. et al. Enhanced cell penetration of acid-degradable particles functionalized with cell-penetrating peptides. Bioconjug. Chem. 19, 876–881 (2008).
Boussif, O. et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethyleneimine. Proc. Natl Acad. Sci. USA 92, 7297–7301 (1995).
Hu, Y. et al. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core–shell nanoparticles. Nano Lett. 7, 3056–3064 (2007).
Hu, Y. et al. Cytosolic delivery mediated via electrostatic surface binding of protein, virus, or siRNA cargos to pH-responsive core–shell gel particles. Biomacromolecules 13, 756–765 (2009).
Verma, A. et al. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nature Mater. 7, 588–595 (2008). This paper suggests that organization of charge and hydrophobicity on nanoparticle surfaces on the single-nanometre scale can allow access to the cytoplasm without endocytosis.
Li, H., Nookala, S. & Re, F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1β and IL-18 release. J. Immunol. 178, 5271–5276 (2007).
Franchi, L. & Nunez, G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1β secretion but dispensable for adjuvant activity. Eur. J. Immunol. 38, 2085–2089 (2008).
Sharp, F. A. et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl Acad. Sci. USA 106, 870–875 (2009).
Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl Acad. Sci. USA 103, 4930–4934 (2006).
Kidane, A. & Park, K. Complement activation by PEO-grafted glass surfaces. J. Biomed. Mater. Res. 48, 640–647 (1999).
Tang, L. P., Liu, L. & Elwing, H. B. Complement activation and inflammation triggered by model biomaterial surfaces. J. Biomed. Mater. Res. 41, 333–340 (1998).
Gadjeva, M. et al. The covalent binding reaction of complement component C3. J. Immunol. 161, 985–990 (1998).
Kemper, C. & Atkinson, J. P. T-cell regulation: with complements from innate immunity. Nature Rev. Immunol. 7, 9–18 (2007).
Carroll, M. C. The complement system in regulation of adaptive immunity. Nature Immunol. 5, 981–986 (2004).
Dempsey, P. W., Allison, M. E., Akkaraju, S., Goodnow, C. C. & Fearon, D. T. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271, 348–350 (1996).
Kolla, R. V. et al. Complement C3d conjugation to anthrax protective antigen promotes a rapid, sustained, and protective antibody response. PLoS ONE 2, e1044 (2007).
Villiers, M. B., Villiers, C. L., Laharie, A. M. & Marche, P. N. Amplification of the antibody response by C3b complexed to antigen through an ester link. J. Immunol. 162, 3647–3652 (1999).
Jokiranta, T. S. et al. Structure of complement factor H carboxyl-terminus reveals molecular basis of atypical haemolytic uremic syndrome. EMBO J. 25, 1784–1794 (2006).
Pascual, M., Plastre, O., Montdargent, B., Labarre, D. & Schifferli, J. A. Specific interactions of polystyrene biomaterials with factor D of human complement. Biomaterials 14, 665–670 (1993).
Singh, M. et al. Polylactide-co-glycolide microparticles with surface adsorbed antigens as vaccine delivery systems. Curr. Drug Deliv. 3, 115–120 (2006).
Malyala, P. et al. The potency of the adjuvant, CpG oligos, is enhanced by encapsulation in PLG microparticles. J. Pharm. Sci. 97, 1155–1164 (2008). In this paper, co-delivery, in the same degradable particle, of antigen and TLR ligand is demonstrated to be beneficial relative to separate but simultaneous delivery.
Malyala, P., O'Hagan, D. T. & Singh, M. Enhancing the therapeutic efficacy of CpG oligonucleotides using biodegradable microparticles. Adv. Drug Deliv. Rev. 61, 218–225 (2009).
Schlosser, E. et al. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine 26, 1626–1637 (2008).
Hamdy, S. et al. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine 26, 5046–5057 (2008).
Blander, J. M. & Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440, 808–812 (2006).
Monks, C. R. F., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82–86 (1998).
Mossman, K. D., Campi, G., Groves, J. T. & Dustin, M. L. Altered TCR signaling from geometrically repatterned immunological synapses. Science 310, 1191–1193 (2005).
Doh, J. & Irvine, D. J. Immunological synapse arrays: patterned protein surfaces that modulate immunological synapse structure formation in T cells. Proc. Natl Acad. Sci. USA 103, 5700–5705 (2006). In this paper, surface patterning methods are used to demonstrate the importance of ultrastructural organization of the interface between a T cell and a dendritic cell.
Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 (2001).
Lee, M. S. & Kim, Y. J. Pattern-recognition receptor signaling initiated from extracellular, membrane, and cytoplasmic space. Mol. Cell 23, 1–10 (2007).
Strindelius, L., Filler, M. & Sjoholm, I. Mucosal immunization with purified flagellin from Salmonella induces systemic and mucosal immune responses in C3H/HeJ mice. Vaccine 22, 3797–3808 (2004).
Nempont, C. et al. Deletion of flagellin's hypervariable region abrogates antibody-mediated neutralization and systemic activation of TLR5-dependent immunity. J. Immunol. 181, 2036–2043 (2008).
Salman, H. H., Gamazo, C., Campanero, M. A. & Irache, J. M. Salmonella-like bioadhesive nanoparticles. J. Control Release 106, 1–13 (2005).
Novak, N., Yu, C. F., Bieber, T. & Allam, J. P. Toll-like receptor 7 agonists and skin. Drug News Perspect. 21, 158–165 (2008).
Thomsen, L. L., Topley, P., Daly, M. G., Brett, S. J. & Tite, J. P. Imiquimod and resiquimod in a mouse model: adjuvants for DNA vaccination by particle-mediated immunotherapeutic delivery. Vaccine 22, 1799–1809 (2004).
Vollmer, J. et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 34, 251–262 (2004).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
van Lookeren Campagne, M., Wiesmann, C. & Brown, E. J. Macrophage complement receptors and pathogen clearance. Cell. Microbiol. 9, 2095–2102 (2007).
Liu, F. J. et al. Independent but not synergistic enhancement to the immunogenicity of DNA vaccine expressing HIV-1 gp120 glycoprotein by codon optimization and C3d fusion in a mouse model. Vaccine 22, 1764–1772 (2004).
Villadangos, J. A. & Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo . Nature Rev. Immunol. 7, 543–555 (2007).
Conner, S. D. & Schmid, S. L. Regulated portals of entry into the cell. Nature 422, 37–44 (2003).
Blander, J. M. & Medzhitov, R. On regulation of phagosome maturation and antigen presentation. Nature Immunol. 7, 1029–1035 (2006).
Huang, Y. B., Leobandung, W., Foss, A. & Peppas, N. A. Molecular aspects of muco- and bioadhesion: tethered structures and site-specific surfaces. J. Control Release 65, 63–71 (2000). In this paper, the mechanism by which grafted polymer chains lead to adhesion to mucus is demonstrated to derive from entanglement, providing a basis for understanding stealth behaviour in the airways.
Wang, Y. Y. et al. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that 'slip' through the human mucus barrier. Angew. Chem. Int. Edn Engl. 47, 9726–9729 (2008). In this paper, the mechanistic understanding of ref. 99 is applied to provide vaccine particles that can penetrate mucus, providing an efficient route to mucosal vaccination.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reprints and permissions information is available at http://www.nature.com/reprints. The authors declare no competing financial interests. Correspondence should be addressed to D.J.M. (mooneyd@seas.harvard.edu).
Rights and permissions
About this article
Cite this article
Hubbell, J., Thomas, S. & Swartz, M. Materials engineering for immunomodulation. Nature 462, 449–460 (2009). https://doi.org/10.1038/nature08604
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature08604
This article is cited by
-
In situ label-free X-ray imaging for visualizing the localization of nanomedicines and subcellular architecture in intact single cells
Nature Protocols (2024)
-
Yeast-derived nanoparticles remodel the immunosuppressive microenvironment in tumor and tumor-draining lymph nodes to suppress tumor growth
Nature Communications (2022)
-
Nano-enabled immunomodulation
Nature Nanotechnology (2021)
-
Hyaluronan is a natural and effective immunological adjuvant for protein-based vaccines
Cellular & Molecular Immunology (2021)
-
Melittin-lipid nanoparticles target to lymph nodes and elicit a systemic anti-tumor immune response
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
