
Abstract
Autism spectrum disorders (ASDs) are childhood neurodevelopmental disorders with complex genetic origins1,2,3,4. Previous studies focusing on candidate genes or genomic regions have identified several copy number variations (CNVs) that are associated with an increased risk of ASDs5,6,7,8,9. Here we present the results from a whole-genome CNV study on a cohort of 859 ASD cases and 1,409 healthy children of European ancestry who were genotyped with ∼550,000 single nucleotide polymorphism markers, in an attempt to comprehensively identify CNVs conferring susceptibility to ASDs. Positive findings were evaluated in an independent cohort of 1,336 ASD cases and 1,110 controls of European ancestry. Besides previously reported ASD candidate genes, such as NRXN1 (ref. 10) and CNTN4 (refs 11, 12), several new susceptibility genes encoding neuronal cell-adhesion molecules, including NLGN1 and ASTN2, were enriched with CNVs in ASD cases compared to controls (P = 9.5 × 10-3). Furthermore, CNVs within or surrounding genes involved in the ubiquitin pathways, including UBE3A, PARK2, RFWD2 and FBXO40, were affected by CNVs not observed in controls (P = 3.3 × 10-3). We also identified duplications 55 kilobases upstream of complementary DNA AK123120 (P = 3.6 × 10-6). Although these variants may be individually rare, they target genes involved in neuronal cell-adhesion or ubiquitin degradation, indicating that these two important gene networks expressed within the central nervous system may contribute to the genetic susceptibility of ASD.
Similar content being viewed by others


Main
ASDs, including autism, are neurodevelopmental disorders characterized by impairments in social and communication skills, as well as stereotyped and repetitive behaviours and/or a restricted range of interests. Current prevalence estimates in the United States are 0.1–0.2% for autism and 0.6% for ASDs1,2.
Linkage and candidate gene association studies have implicated several chromosomal regions in autism3,4. However, positive findings in one study often fail to replicate in other studies, and a consistent picture of susceptibility loci in autism is still lacking. Some telling clues about ASD genetics arose from recent studies on CNVs5, including the association of de novo CNVs with ASDs6. Although de novo CNVs that disrupt specific genes may contribute to the pathogenesis of ASDs, heritable CNVs are much more common but have been less studied as risk factors of ASDs. A family-based genome-wide linkage and CNV analysis by the Autism Genome Project Consortium using Affymetrix 10K single nucleotide polymorphism (SNP) arrays implicated chromosome 11p12-13 and neurexin 1 (NRXN1) as candidate loci7. A study using the Affymetrix 500K SNP array in a Canadian population reported 277 rare CNVs that were only observed in ASD patients but not in 1,652 healthy controls or in the Database of Genomic Variants8. Furthermore, 16p11.2 deletions and duplications have been reported in independent cohorts of autism patients9. These studies concordantly implicate a role for CNVs in the genetic susceptibility to ASDs.
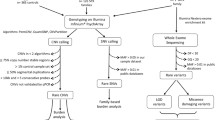
To search systematically for CNVs that confer a risk to ASDs, we used a genome-wide approach with the Illumina HumanHap550 BeadChip. We assembled an Autism Case-Control (ACC) cohort by collecting 859 ASD cases (from a total of 1,246 ACC cases, parents, and siblings) of European ancestry, and 1,409 healthy controls. Among these case subjects, all met the diagnostic criteria for autism on the basis of Autism Diagnostic Interview (ADI), and 124 met the criteria for other ASDs on the basis of Autism Diagnostic Observation Schedule (ADOS)13. Fifty-four per cent were from simplex families, the rest were from multiplex families. In addition, we also analysed 1,336 ASD cases (from a total of 3,398 cases, parents, and siblings) in the Autism Genetic Resource Exchange (AGRE)14 collection, as well as 1,110 control subjects as a replication cohort. Among the AGRE cases, 5% were from simplex families and 95% were from multiplex families: 1,202 met the criteria for autism and 134 for other ASDs13 (Supplementary Tables 1 and 2).
We generated 78,490 CNV calls (22,581 in the ACC series and 55,909 in the AGRE series) from all the ASD subjects and their family members that met strictly established data quality thresholds (Methods). On average, 15.5 CNV calls were made for each individual using the PennCNV software15, with similar frequency observed in cases and controls (Supplementary Fig. 2).
We first examined eight genomic regions that have been previously implicated in ASDs. Among those, CNVs involving the 15q11-13, 22q11.21, and NRXN1 regions have well-established associations with autism10. CNVs affecting CNTN4 in ASD cases have also been reported in independent studies11,12. We statistically adjusted for relatedness of cases with permutation and our results demonstrate that duplications of 15q11-q13 and the 22q11.21 region, deletions of NRXN1, as well as deletions and duplications of CNTN4 replicate in our cohorts (Table 1). Conversely, we did not obtain statistical support for several other genomic regions previously shown to associate with ASD, including AUTS2 (ref. 16), NLGN3 (ref. 17), SHANK3 (ref. 18) and 16p11.2 (ref. 9) (Table 1). We observed a similar frequency of deletions and duplications of the 16p11.2 locus in the ASD cases (∼0.3%) as previously reported9; however, the CNV frequency in the control subjects at this locus was also comparable to that of the cases (Supplementary Fig. 3). It is noteworthy that CNVs at the 16p11.2 locus do not segregate to all cases in three of the affected families and they are also transmitted to unaffected siblings (Supplementary Fig. 4). These results indicate that CNVs at the 16p11.2 locus may not be sufficient to be causal variants in ASD.
To identify other new genomic loci contributing to ASDs, we applied a segment-based scoring approach that scans the genome for consecutive SNPs with more frequent copy number changes in cases compared to controls. This approach defines copy number variation regions, or CNVRs (Supplementary Fig. 5, upper panel). In the ACC cohort, we identified four CNVRs that were observed in cases but not in controls, as well as five CNVRs that had significantly higher frequency in cases versus controls (Table 2).
To replicate the CNVRs exclusively observed in ACC cases, we examined the AGRE case-control data set; of the four case-specific CNVRs, two were also exclusive to AGRE cases (PARK2 and RFWD2), whereas the other two (AK057321 and FBXO40) were not observed in either the cases or controls (combined P values ranging from 3.57 × 10-6 to 0.1, unadjusted for multiple testing) (Table 2). Interestingly, four genes (UBE3A, PARK2, RFWD2 and FBXO40) that were significantly enriched for CNVs and observed in the ASD cases only, belong to the ubiquitin gene family (UniProt category ‘Ubl conjugation pathway’, P = 3.3 × 10-3). The other five CNVRs, as well as being enriched in the ACC cases compared with controls, were over-represented in the AGRE cases compared with the independent controls (Table 2). Figure 1 shows the most statistically significant locus, a duplication 55 kb upstream of AK123120, using UCSC Genome Browser19 with Build 36 (March 2006) of the human genome. To ensure reliability of our CNV detection method, we experimentally validated all the significant CNVRs using other methods, including quantitative PCR (qPCR) and multiplex ligation-dependent probe amplification (MLPA) (Fig. 2). Affymetrix 5.0 array data were also available for a subset of the AGRE subjects for validation.

AK123120 chromosome 2 (chr2): 12,986,750–13,291,000 divided into subsections with headers for ACC CNVs, AGRE CNVs, AGRE Affymetrix validation CNVs, and control CNVs. The AGRE Affymetrix Replication track is on the basis of genome-wide 5.0 SNP genotyping data from the Broad Institute (see Supplementary Methods and Acknowledgements), and were generated using the PennCNV-Affy algorithm (see Supplementary Methods), to serve as a further means to validate the Illumina-based CNV calls. SNP and copy number (CN) probe coverage are shown as blue lines across the top. Produced with custom tracks listing CNV calls uploaded to http://genome.ucsc.edu. Figures for all loci are included in Supplementary Information.

Fluorescent probe-based qPCR assays using Roche Universal probe library and/or MLPA were designed to validate every candidate CNV with a completely independent test (representative series shown for each locus). Error bars denote the s.d. of quadruplicate runs. bp, base pairs; Del, deletion; Dup, duplication.
Besides a segment-based scoring approach for CNV association, an alternative method is the gene-based scoring approach that examines CNV calls affecting any region of the gene (Supplementary Fig. 5, lower panel). Using this approach, we further identified seven genes with an increased frequency of CNVs in ASD cases versus controls (Supplementary Table 3). For each gene, most CNVs target different parts of the gene and would have been missed by the segment-based approach. Of note, four of the genes identified by the segment- and gene-based approaches are involved in neuron development (NRXN1, CNTN4, ASTN2 and NLGN1) (Gene Ontology term ‘neuron development’, P = 9.5 × 10-3). Therefore, by combining evidence from two complementary CNV association approaches, the large sample size has enabled us to implicate two specific gene networks or biological pathways in ASDs: the ubiquitination system and neuronal cell-adhesion molecules.
The genes from the ubiquitin pathway (UBE3A, PARK2, RFWD2 and FBXO40) represent a new CNV finding in ASD susceptibility. Ubiquitination is a post-translational modification which can rapidly alter protein function and target proteins for proteasome-mediated degradation. The ubiquitin–proteasome system operates pre- and post-synaptic compartments, regulating synaptic attributes, including neurotransmitter release, synaptic vesicle recycling in presynaptic terminals, and dynamic changes in dendritic spines and the post-synaptic density (PSD)20. Of the four ubiquitin-related genes highlighted in our study, UBE3A, a ubiquitin protein ligase, has been the most extensively studied in the context of autism. PARK2 is a ubiquitin-protein ligase, mutations of which cause autosomal recessive juvenile Parkinson’s disease21, and RFWD2 and FBXO40 are also ubiquitin-protein ligases, but neither has been previously associated with disease-causing mutations. The role of ubiquitin in the turnover of synaptic components such as the neuronal cell-adhesion molecules in a process involving regulation of activity-dependent synaptic plasticity presents a mechanism that links these two major gene networks. In addition to the genes described above, several ubiquitin-related genes are involved in human neurological diseases. These include NHLRC1, UBR, CUL4B, BRWD3 and HUWE1, genes that encode ubiquitin protein E3 ligases. Mutations in the latter three and in UBE2A, an E2 ubiquitin-conjugating enzyme, cause syndromes that include intellectual disability22.
Genes from the second group of genes implicated in our study, neuronal cell-adhesion molecules, are critical in the development of the nervous system, contributing to axonal guidance, synaptic formation and plasticity, and neuronal–glial interactions. Recent genetic evidence has suggested associations between autism susceptibility and neuronal cell-adhesion molecules, including NRXN1 (ref. 10), CNTNAP2 (ref. 23), NLGN3 (ref. 17 ), NLGN4X (ref. 17), and specific cadherins. Our results provide support for some previously reported genes (NRXN1 and CNTN4), and also implicate additional genes with cell-adhesion functions, including NLGN1 and ASTN2. Mutations in neuroligin superfamily members have previously been found in individuals with autism, and have subsequently been shown to be functionally relevant24. ASTN1, a well-studied homologue of ASTN2, is a neuronal protein receptor integral in the process of glial-guided granule cell migration during development25, and ASTN2 deletions have recently been associated with schizophrenia26.
Using a genome-wide approach for high-resolution CNV detection, we have identified candidate genomic loci with enrichment of CNVs in ASD cases as compared to controls, and have replicated many of them using an independent set of cases. Most of these genes fall within two pathways/networks involving neuronal cell-adhesion and ubiquitin degradation. The enrichment of genes within these molecular systems suggests new susceptibility mechanisms for ASDs. Our results call for functional and expression assays to be completed to assess the biological effects of CNVs in these candidate genes.
Methods Summary
All genome-wide SNP genotyping was performed using the InfiniumII HumanHap550 BeadChip at the Center for Applied Genomics at The Children’s Hospital of Philadelphia (CHOP). We called CNVs with the PennCNV algorithm15, which combines multiple values, including Log R Ratio, B Allele Frequency, SNP spacing and population frequency of the B allele into a hidden Markov model. The term ‘CNV’ represents individual CNV calls, whereas ‘CNVR’ refers to population-level variation. Quality control thresholds included a high success rate of attempted SNPs, low standard deviation of normalized intensity, genetically inferred European ancestry, low genomic wave artefacts, count of CNV calls per subject, and genotypic duplicate removal (Supplementary Table 4). CNV frequency between cases and controls was evaluated at each SNP using Fisher’s exact test. We report statistical local minimums in reference to a region of nominal significance including SNPs residing within 1 Mb of each other (Supplementary Fig. 6). Resulting significant CNVRs were excluded if they were (1) residing on telomere or centromere proximal cytobands; (2) arising from a ‘peninsula’ of common CNV (Supplementary Fig. 7); (3) genomic regions with extremes in GC content27; or (4) samples contributing to multiple CNVRs. To adjust for siblings in the AGRE data, we calculated a permutation-based P value (×1,000), in which disease labels for siblings were permutated together. DAVID (Database for Annotation, Visualization, and Integrated Discovery)28 assessed the significance of functional annotation clustering. We considered loci significant between cases and controls (P < 0.05) where ACC discovery cases had overlapping variation, replicated in AGRE or were not observed in control subjects, and validated with another method (qPCR Roche Universal Probe Library using qBase29, MRC-Holland MLPA, and Affymetrix 5.0 from Broad). Statistical correction of five deletion and nine duplication CNVRs, on the basis of discovery cohort (ACC) significance and signal review is appropriate for our study (‘CNV Filtering Steps’ in Supplementary Materials).
References
Autism and Developmental Disabilities Monitoring Network. 〈http://www.cdc.gov/mmwr/pdf/ss/ss5601.pdf〉 (2007)
Newschaffer, C. J. et al. The epidemiology of autism spectrum disorders. Annu. Rev. Public Health 28, 235–258 (2007)
Gupta, A. R. & State, M. W. Recent advances in the genetics of autism. Biol. Psychiatry 61, 429–437 (2007)
Klauck, S. M. Genetics of autism spectrum disorder. Eur. J. Hum. Genet. 14, 714–720 (2006)
Vorstman, J. A. S. et al. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry 11, 18–28 (2006)
Sebat, J. et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007)
Szatmari, P. et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature Genet. 39, 319–328 (2007)
Marshall, C. R. et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488 (2008)
Weiss, L. A. et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675 (2008)
Kim, H. G. et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207 (2008)
Roohi J. et al. Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 46, 176–182 (2008)
Fernandez, T. et al. Disruption of Contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 82, 1385 (2008)
Le Couteur, A. et al. Diagnosing autism spectrum disorders in pre-school children using two standardised assessment instruments: the ADI-R and the ADOS. J. Autism Dev. Disord. 38, 362–372 (2008)
Geschwind, D. H. et al. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am. J. Hum. Genet. 69, 463–466 (2001)
Wang, K. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674 (2007)
Kalscheuer, V. M. et al. Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 121, 501–509 (2007)
Jamain, S. et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature Genet. 34, 27–29 (2003)
Moessner, R. et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297 (2007)
Kent W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002)
Yi, J. J. & Ehlers, M. D. Ubiquitin and protein turnover in synapse function. Neuron 47, 629–632 (2005)
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998)
Tai, H.-C. & Schuman, E. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nature Rev. Neurosci. 9, 826–838 (2008)
Alarcón, M. et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159 (2008)
Chubykin, A. A. et al. Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J. Biol. Chem. 280, 22365–22374 (2005)
Zheng, C., Heintz, N. & Hatten, M. E. CNS gene encoding astrotactin, which supports neuronal migration along glial fibers. Science 272, 417–419 (1996)
Kahler, A. K. et al. Association analysis of schizophrenia on 18 genes involved in neuronal migration: MDGA1 as a new susceptibility gene. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 147B, 1089–1100 (2008)
Diskin, S. et al. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 36, e126 (2008)
Dennis, G. et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, R60 (2003)
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F. & Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19 (2007)
Acknowledgements
We gratefully thank all the ASD children and their families at the participating study sites who were enrolled in this study and all the control subjects who donated blood samples to Children’s Hospital of Philadelphia for genetic research purposes. We thank the technical staff in the Center for Applied Genomics, Children’s Hospital of Philadelphia for generating the genotypes used in this study. We thank S. Diskin for her contribution to the discussion on the effect of wave artifacts on CNV calling. We also thank S. Kristinsson, L. A. Hermannsson and A. Krisbjörnsson for their software design and contribution. This research was financially supported by The Children’s Hospital of Philadelphia, Autism Speaks, and NICHD (HD35476). We also gratefully acknowledge the resources provided by the AGRE Consortium (D. H. Geschwind, M. Bucan, W. T. Brown, J. D. Buxbaum, R. M. Cantor, J. N. Constantino, T. C. Gilliam, C. M. Lajonchere, D. H. Ledbetter, C. Lese-Martin, J. Miller, S. F. Nelson, G. D. Schellenberg, C. A. Samango-Sprouse, S. Spence, M. State, R. E. Tanzi) and the participating families. AGRE is a program of Autism Speaks and is at present supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to C. M. Lajonchere (PI), and formerly by grant MH64547 to D. H. Geschwind (PI). The AGRE data set was genotyped by the Center for Applied Genomics at the Children’s Hospital of Philadelphia, which funded all genotyping for this study, and the complete sets of genotyping data have been released to the public domain. AGRE-approved academic researchers can acquire the data sets from AGRE (http://www.agre.org). The study is supported in part by Research Award from the Margaret Q. Landenberger Foundation (H.H.), UL1-RR024134-03 (H.H.), a Research Development Award from the Cotswold Foundation (H.H. and S.F.A.G.), the Beatrice and Stanley A. Seaver Foundation (J.D.B.), the Department of Veterans Affairs (G.D.S.), and National Institute of Health grants HD055782-01 (J.Munson, A.E., O.K., G.D. and G.D.S.), MH0666730 (J.D.B.), MH061009 and NS049261 (J.S.S.), HD055751 (E.H.C.), U10MH66766-02S1 (C.J.M.), MH69359, M01-RR00064 and the Utah Autism Foundation (H.C., J.Miller, and W.M.M.). All genotyping for this study was supported by an Institutional Development Award to the Center for Applied Genomics (H.H.) from the Children’s Hospital of Philadelphia. We acknowledge the Autism Genome Project Consortium (J.P., C.W.B., T.H.W., W.M.M., H.C., J.I.N., J.S.S., E.H.C., J.Munson, A.E., O.K., J.D.B., B.D. and G.D.S.) funded by Autism Speaks, the Medical Research Council (UK) and the Health Research Board (Ireland). We are grateful for the public access to the Affymetrix 5.0 data available for a subset of AGRE families (http://www.agre.org), which served as important validation of CNV calls made.
Author Contributions H.H. and G.D.S. designed the study and supervised the data analysis and interpretation. J.T.G., K.W. and B.D. conducted the statistical analyses. C.E.K. and E.C.F. directed the genotyping of stage 1. J.D.B. coordinated the validation. G.C. and O.K. performed qPCR and MLPA validation of CNVs and edited the manuscript. J.T.G., K.W., H.H., G.D.S. and B.D. drafted the manuscript. G.D.S., N.J.M., E.H.C., W.M.M., H.C., T.H.W., J.D.B., T.O., J.I.N., E.A., L.S., J.R., T.S., C.B., C.J.M., D.J.P. and D.Z. collected samples and contributed phenotype data for the study and assisted with data collection and manuscript preparation. E.C., S.F.A.G., P.S., M.I., B.D., L.K., S.W. and K.W. reviewed the data, assisted with interpretation of the data, and edited the manuscript. Other authors contributed to sample acquisition and processing or to data analysis and interpretation.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Figures 1-9 with Legends, Supplementary Materials, Supplementary Tables 1-4 and Supplementary Data. (PDF 2623 kb)
PowerPoint slides
Rights and permissions
About this article
Cite this article
Glessner, J., Wang, K., Cai, G. et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 (2009). https://doi.org/10.1038/nature07953
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature07953
This article is cited by
-
Prenatal Programming of Monocyte Chemotactic Protein-1 Signaling in Autism Susceptibility
Molecular Neurobiology (2024)
-
Neurexin1α knockout in rats causes aberrant social behaviour: relevance for autism and schizophrenia
Psychopharmacology (2024)
-
GRID1/GluD1 homozygous variants linked to intellectual disability and spastic paraplegia impair mGlu1/5 receptor signaling and excitatory synapses
Molecular Psychiatry (2024)
-
Removal of a partial genomic duplication restores synaptic transmission and behavior in the MyosinVA mutant mouse Flailer
BMC Biology (2023)
-
Rare recurrent copy number variations in metabotropic glutamate receptor interacting genes in children with neurodevelopmental disorders
Journal of Neurodevelopmental Disorders (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
