
Abstract
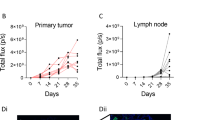
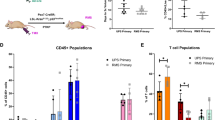
A fundamental question in cancer biology is whether cells with tumorigenic potential are common or rare within human cancers. Studies on diverse cancers, including melanoma, have indicated that only rare human cancer cells (0.1–0.0001%) form tumours when transplanted into non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice. However, the extent to which NOD/SCID mice underestimate the frequency of tumorigenic human cancer cells has been uncertain. Here we show that modified xenotransplantation assay conditions, including the use of more highly immunocompromised NOD/SCID interleukin-2 receptor gamma chain null (Il2rg-/-) mice, can increase the detection of tumorigenic melanoma cells by several orders of magnitude. In limiting dilution assays, approximately 25% of unselected melanoma cells from 12 different patients, including cells from primary and metastatic melanomas obtained directly from patients, formed tumours under these more permissive conditions. In single-cell transplants, an average of 27% of unselected melanoma cells from four different patients formed tumours. Modifications to xenotransplantation assays can therefore dramatically increase the detectable frequency of tumorigenic cells, demonstrating that they are common in some human cancers.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
References
Schatton, T. et al. Identification of cells initiating human melanomas. Nature 451, 345–349 (2008)
Li, C. et al. Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037 (2007)
Prince, M. E. et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl Acad. Sci. USA 104, 973–978 (2007)
Wu, C. et al. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 67, 8216–8222 (2007)
Wang, J. C. et al. High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase. Blood 91, 2406–2414 (1998)
Al-Hajj, M. et al. Prospective identification of tumorigenic breast cancer cells. Proc. Natl Acad. Sci. USA 100, 3983–3988 (2003)
Cox, C. V. et al. Characterization of acute lymphoblastic leukemia progenitor cells. Blood 104, 2919–2925 (2004)
Hope, K. J., Jin, L. & Dick, J. E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nature Immunol. 5, 738–743 (2004)
Dalerba, P. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl Acad. Sci. USA 104, 10158–10163 (2007)
O’Brien, C. A. et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110 (2007)
Ricci-Vitiani, L. et al. Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115 (2007)
Williams, R. T., den Besten, W. & Sherr, C. J. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 21, 2283–2287 (2007)
Kelly, P. N. et al. Tumor growth need not be driven by rare cancer stem cells. Science 317, 337 (2007)
Somervaille, T. C. & Cleary, M. L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 10, 257–268 (2006)
Agliano, A. et al. Human acute leukemia cells injected in NOD/LtSz-SCID/IL-γnull mice generate a faster and more efficient disease compared to other NOD/SCID-related strains. Int. J. Cancer 123, 2222–2227 (2008)
Feuring-Buske, M. et al. Improved engraftment of human acute myeloid leukemia progenitor cells in β-2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia 17, 760–763 (2003)
Kennedy, J. A. et al. Comment on ‘Tumor growth need not be driven by rare cancer stem cells’. Science 318, 1722; author reply 1722 (2007)
Pardal, R., Clarke, M. F. & Morrison, S. J. Applying the principles of stem-cell biology to cancer. Nature Rev. Cancer 3, 895–902 (2003)
Wang, J. C. & Dick, J. E. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 15, 494–501 (2005)
Shackleton, M. et al. Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88 (2006)
Shultz, L. D. et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγnull mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489 (2005)
Ito, M. et al. NOD/SCID/γc null mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100, 3175–3182 (2002)
McKenzie, J. L. et al. Human short-term repopulating stem cells are efficiently detected following intrafemoral transplantation into NOD/SCID recipients depleted of CD122+ cells. Blood 106, 1259–1261 (2005)
Shultz, L. D. et al. Regulation of human short-term repopulating cell (STRC) engraftment in NOD/SCID mice by host CD122+ cells. Exp. Hematol. 31, 551–558 (2003)
Hamada, K. et al. Liver metastasis models of colon cancer for evaluation of drug efficacy using NOD/Shi-scid IL2Rγnull (NOG) mice. Int. J. Oncol. 32, 153–159 (2008)
Suemizu, H. et al. Identification of a key molecular regulator of liver metastasis in human pancreatic carcinoma using a novel quantitative model of metastasis in NOD/SCID/γcnull (NOG) mice. Int. J. Oncol. 31, 741–751 (2007)
Ishikawa, F. et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature Biotechnol. 25, 1315–1321 (2007)
Kleinman, H. K. & Martin, G. R. Matrigel: basement membrane matrix with biological activity. Semin. Cancer Biol. 15, 378–386 (2005)
Pretlow, T. G. et al. Transplantation of human prostatic carcinoma into nude mice in Matrigel. Cancer Res. 51, 3814–3817 (1991)
Sweeney, T. M. et al. Basement membrane and the SIKVAV laminin-derived peptide promote tumor growth and metastases. Cancer Metastasis Rev. 10, 245–254 (1991)
Fridman, R. et al. Enhanced tumor growth of both primary and established human and murine tumor cells in athymic mice after coinjection with Matrigel. J. Natl Cancer Inst. 83, 769–774 (1991)
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004)
Iwashita, T. et al. Hirschsprung disease is linked to defects in neural crest stem cell function. Science 301, 972–976 (2003)
Bao, S. et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 68, 6043–6048 (2008)
Frank, N. Y. et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 65, 4320–4333 (2005)
Yilmaz, O. H. et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 (2006)
Horwich, A., Shipley, J. & Huddart, R. Testicular germ-cell cancer. Lancet 367, 754–765 (2006)
Kleinsmith, L. J. & Pierce, G. B. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 24, 1544–1551 (1964)
Bonnefoix, T. et al. Fitting limiting dilution experiments with generalized linear models results in a test of the single-hit Poisson assumption. J. Immunol. Methods 194, 113–119 (1996)
Nakajima, T. et al. Immunohistochemical demonstration of S100 protein in malignant melanoma and pigmented nevus, and its diagnostic application. Cancer 50, 912–918 (1982)
Fernandez, Y. et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 65, 6294–6304 (2005)
Wang, Z. et al. Ablation of proliferating marrow with 5-fluorouracil allows partial purification of mesenchymal stem cells. Stem Cells 24, 1573–1582 (2006)
Bancroft, J. D. & Stevens, A. Theory and Practice of Histological Techniques (Churchill Livingstone, 1990)
Acknowledgements
This work was supported by the Howard Hughes Medical Institute and by the Allen H. Blondy Research Fellowship. The University of Michigan Melanoma Bank was supported by a gift from Lewis and Lillian Becker. Flow cytometry was partly supported by the University of Michigan Comprehensive Cancer Center grant from the National Institutes of Health CA46592. We thank: D. Adams, M. White and the University of Michigan Flow Cytometry Core Facility for support; N. McAnsh and the University of Michigan Cancer Centre Histology Core for histological studies; G. K. Smyth for assistance with statistics; and Z. Azizan for support with tissue collection. Antibody production was supported in part by the National Institute of Diabetes, Digestive, and Kidney Diseases, grant NIH5P60-DK20572 to the Michigan Diabetes Research and Training Center. Some antibodies were provided by Caltag or by eBioscience to screen for cancer stem-cell markers. Human primary melanocyte cultures were provided by M. Soengas. Human mesenchymal stem cells were provided by Z. Wang and P. Krebsbach. E.Q. was supported by the Spanish Ministry of Education and the Marie Curie Outgoing International Fellowship from the European Commission. M.S. was supported by the Australian National Health and Medical Research Council, the Human Frontiers Science Program and Australia Post.
Author Contributions E.Q., M.S. and S.J.M. planned the project. E.Q. and M.S. performed the experiments, and analysed data with S.J.M. M.S.S. and T.M.J. obtained consent from the patients and surgically obtained many of the melanoma specimens. D.R.F. performed all pathology and diagnosed the tumours with T.M.J. T.M.J. banked the melanomas, and provided clinical information. E.Q., M.S. and S.J.M. wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1, 2 and Supplementary Figures 1-6 with legends. (PDF 3549 kb)
Rights and permissions
About this article
Cite this article
Quintana, E., Shackleton, M., Sabel, M. et al. Efficient tumour formation by single human melanoma cells. Nature 456, 593–598 (2008). https://doi.org/10.1038/nature07567
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature07567
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
