
Abstract
The reverse transcriptase of human immunodeficiency virus (HIV) catalyses a series of reactions to convert the single-stranded RNA genome of HIV into double-stranded DNA for host-cell integration. This task requires the reverse transcriptase to discriminate a variety of nucleic-acid substrates such that active sites of the enzyme are correctly positioned to support one of three catalytic functions: RNA-directed DNA synthesis, DNA-directed DNA synthesis and DNA-directed RNA hydrolysis. However, the mechanism by which substrates regulate reverse transcriptase activities remains unclear. Here we report distinct orientational dynamics of reverse transcriptase observed on different substrates with a single-molecule assay. The enzyme adopted opposite binding orientations on duplexes containing DNA or RNA primers, directing its DNA synthesis or RNA hydrolysis activity, respectively. On duplexes containing the unique polypurine RNA primers for plus-strand DNA synthesis, the enzyme can rapidly switch between the two orientations. The switching kinetics were regulated by cognate nucleotides and non-nucleoside reverse transcriptase inhibitors, a major class of anti-HIV drugs. These results indicate that the activities of reverse transcriptase are determined by its binding orientation on substrates.
Similar content being viewed by others


Main
Virtually all RNA-processing and DNA-processing enzymes show selectivity for backbone compositions or base sequences of their nucleic-acid substrates. This substrate selectivity is especially crucial for the HIV-1 reverse transcriptase (RT), which binds and discriminates between a variety of nucleic-acid duplexes for distinct catalytic functions1,2. RT is a heterodimer consisting of a p51 and a p66 subunit, the latter of which contains catalytically active DNA polymerase and RNase H domains3,4, catalysing a complex, multi-step reaction to convert the single-stranded RNA genome into double-stranded DNA1,2. First, RT uses the viral RNA genome as a template and a host-cell transfer RNA as a primer to synthesize a minus-strand DNA, producing an RNA–DNA hybrid5,6,7. This duplex becomes the substrate of the RNase H domain of RT, which cleaves the RNA strand at numerous points, leaving behind short RNA segments hybridized to the nascent DNA8,9,10. Among these RNAs, two specific purine-rich sequences, known as the polypurine tracts (PPTs), serve as unique primers to initiate the synthesis of plus-strand DNA11,12,13, thereby creating the double-stranded DNA viral genome. Specific cleavage by RNase H then removes the PPT primers and exposes the integration sequence to facilitate the insertion of the viral DNA into the host chromosome14. Inappropriate initiation of synthesis of the plus-strand DNA at other RNA segments prevents integration2,15. RT must therefore obey the following primer-selection rules: first, DNA primers readily engage the polymerase activity of RT; second, generic RNA primers are not efficiently extended by RT but readily engage the RNase H activity of RT when annealed with DNA; third, the PPT RNA can direct both the DNA polymerase activity and a site-specific RNase H activity of RT. The mechanism by which RT discriminates between these substrates and executes the appropriate catalytic function is, however, poorly understood. Although RNase H cleavage analysis suggests the presence of different interaction modes of RT with substrates16,17, crystal structures have so far revealed only one enzyme-binding orientation4,18,19,20,21,22.
Single-molecule assay for enzyme–substrate interactions
For a better understanding of how RT interacts with substrates, we designed a single-molecule assay to measure the enzyme orientation relative to its substrate by using fluorescence resonance energy transfer (FRET)23,24, a method well suited for probing dynamic interactions between proteins and nucleic acids25,26,27. Static FRET measurements have also been used previously to characterize the pre-translocation and post-translocation states of RT on a DNA duplex28. Because RT accommodates 19–22 base pairs of nucleic-acid duplex within its primer–template-binding cleft19,22,29 (Fig. 1a), we constructed several duplex substrates with different backbone compositions and base sequences, each consisting of a 50-nucleotide (nt) oligonucleotide mimicking the template and a complementary 19–21-nt oligonucleotide emulating the primer (Fig. 1b and Supplementary Fig. 1). A Cy5 fluorophore was specifically attached to one of the single-stranded overhang regions on the template to serve as the FRET acceptor. We refer to the labelling schemes with Cy5 near the 5′ and 3′ ends of the primer as the 5* and 3* labels, respectively.

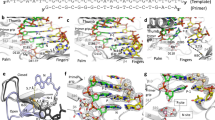
a, The structure of HIV-1 RT bound to a DNA–DNA substrate21. Labelling sites for Cy3 on RT are highlighted by green stars. b, Nucleic-acid substrates consisted of a 19–21-nt primer strand annealed to a 50-nt template strand containing a Cy5 label (red star). Cy5 was either 3 nt from the 5′ end (circle) or 4–6 nt from the 3′ end (arrow) of the primer. c, Single-molecule detection of Cy3 (green star or sphere)-labelled RT binding to and dissociating from the surface-immobilized nucleic-acid substrates labelled with Cy5 (red star or sphere). The stars and spheres indicate dyes that do and do not emit fluorescence, respectively. d, FRET analysis for RT binding to a single primer–template complex. Top: fluorescence time traces from Cy3 (green) and Cy5 (red) under excitation at 532 nm and that from Cy5 (pink) under excitation at 635 nm. Middle: FRET value calculated over the duration of the binding events (yellow shaded regions). Bottom: FRET distribution histogram created for the binding events.
Surface-immobilized substrates were immersed in a solution containing RT molecules labelled with a FRET donor dye, Cy3, either at the RNase H domain (H-labelled) or at the fingers domain (F-labelled) of the p66 subunit (Fig. 1a). The two dye-labelling sites were located on opposite poles of the enzyme and separated by about 8 nm. An E478Q mutation was introduced into the RNase H domain to abolish its RNA cleavage activity so as to prevent the RT-induced degradation of nucleic-acid substrates during observation30. Experiments were conducted with this RNase-H-inactive variant unless otherwise mentioned. Neither dye attachment nor surface immobilization significantly altered the polymerase activity of RT (Supplementary Fig. 2).
Fluorescence of individual duplex substrates on the slide was monitored by using total-internal-reflection fluorescence (TIRF) microscopy with alternating laser excitations25 at 532 and 635 nm (Fig. 1c). The 532-nm light excites the FRET donor Cy3 without significantly exciting the acceptor Cy5, allowing us to detect the FRET between the Cy3-labelled RT and the Cy5-labelled substrate. The 635-nm light directly excites the Cy5 dye, providing a means to probe the presence of the nucleic-acid substrate and FRET acceptor independently of RT binding. Freely diffusing RT was observed to bind and dissociate from the substrates in real time. Each binding event caused an increase in the total fluorescence signal collected from both Cy3 and Cy5 channels under the 532-nm excitation without affecting the signal obtained under the 635-nm excitation (Fig. 1d). The observed FRET value allowed the enzyme orientation of each binding event to be determined.
RT binds DNA and RNA primers in opposite orientations
We first examined the binding orientation of RT on a 19-nt DNA primer hybridized to a 50-nt DNA template. When H-labelled RT was added to the 5*-labelled substrates, binding events consistently yielded high FRET values (centred at about 0.94; Fig. 2a), indicating an overwhelming tendency for the enzyme to bind with its RNase H domain close to the 5′ terminus of the primer. Conversely, predominantly low FRET values (about 0.14) were observed when H-labelled RT bound to an isogenic 3*-labelled substrate (Fig. 2b). Furthermore, F-labelled RT bound primarily to the 3*-labelled substrate with high FRET values (about 0.90; Fig. 2c), indicating that the DNA polymerase domain was located near the 3′ end of the primer. As a control, when both Cy3 and Cy5 were placed on the substrates, either near the same end of or flanking the duplex region, no significant change in FRET was observed on the addition of RT, suggesting that RT does not cause a sizable change in the photophysical properties of the dyes or the end-to-end distance of the duplex (Supplementary Fig. 3). Taken together, these results indicate that RT binds to the DNA–DNA primer–template complex with its polymerase active site between the fingers and palm domains close to the 3′ end of the primer and the RNase H domain near the 5′ end—an orientation that matches the polymerization-competent binding mode observed in RT–substrate co-crystal structures19,20,21,22. A virtually identical binding orientation was observed for RT on a 19-nt DNA primer annealed to a 50-nt RNA template (Supplementary Fig. 4). The same binding orientation was also observed for the RNase-H-active RT (without the E478Q mutation) on the DNA–DNA primer–template complex (Supplementary Fig. 5a).

a, RT with Cy3 (green star) attached in the H-labelling scheme was allowed to bind substrates consisting of DNA primer and template (black arrows), with Cy5 (red star) attached in the 5*-labelling scheme. b, H-labelled RT bound to 3*-labelled DNA duplex substrates. c, F-labelled RT bound to 3*-labelled DNA duplex substrates. d, H-labelled RT bound to 5*-labelled hybrid duplex substrates consisting of a RNA primer (orange arrow) and a DNA template (black arrow). e, H-labelled RT bound to 3*-labelled hybrid duplex substrates. f, F-labelled RT bound to 5*-labelled hybrid duplex substrates. The RT binding orientations consistent with the FRET distributions are depicted.
Next we examined binding to an RNA primer annealed to a DNA template. The primer and template sequences were identical to those used above. Again, RT adopted a predominantly single binding configuration, but now with a drastically different orientation: H-labelled RT bound to the 5*-labelled substrates with low FRET values (about 0.27; Fig. 2d) but to the 3*-labelled substrates with primarily high FRET values (about 0.95; Fig. 2e); F-labelled RT bound to 5*-labelled substrate with high FRET values (about 0.88; Fig. 2f). These results unambiguously define a binding orientation on the RNA primer that is opposite to that on the DNA primer, with the DNA polymerase domain adjacent to the 5′ terminus of the primer and the RNase H domain close to the 3′ end. The same binding orientation was also found for the RNase-H-active RT (Supplementary Fig. 5b). This orientation clearly cannot support primer extension activity but directly explains the primary RNase-H-cleavage mode observed previously on similar substrates, in which the cleavage site is 18 nt from the 5′ terminus of the RNA17. The two opposite binding orientations on DNA and RNA primers were also observed on primers encoding an alternative sequence (Supplementary Fig. 6).
To identify which features were most important in discriminating between DNA and RNA primers and directing RT orientation, we designed a series of 19-nt chimaeric primers containing different compositions of RNA and DNA nucleotides denoted by xR:yD (that is, x RNA nt at the 5′ end and y DNA nt at the 3′ end). These chimaeras were annealed to a 50-nt DNA template and incubated with H-labelled RT (Fig. 3a and Supplementary Fig. 7). Whereas pure DNA and RNA primers bound to RT predominantly in a single orientation, most chimaeric primers supported both high and low FRET orientations (Supplementary Fig. 7). The free-energy difference (ΔG) between the two states was most sensitive to the sugar composition of the four or five nucleotides located at each end of the 19-nt primer (Fig. 3b), suggesting that the interactions between RT and nucleic acid at opposite ends of the primer–template binding cleft were most important in determining the binding orientation. This observation is consistent with the crystal structures, which show RT–substrate contacts primarily clustered in two regions near the DNA polymerase and RNase H active sites19,22. A single nucleotide provided the strongest determinant of binding orientation: changing the sugar content of the fifth nucleotide from the primer 5′ terminus alone caused a nearly 2kBT (where kB is the Boltzmann constant) change in ΔG (Fig. 3b). This position makes specific contacts with RT residues T473 and Q475 located within the RNase H primer grip19,22. These residues are conserved between RNases H found in viruses, bacteria and humans22,31,32. Replacement of these residues with alanine in HIV-1 RT decreases the DNA synthesis rate and inhibits virus infectivity33. Overall, the backbone composition of nucleotides near the 5′ end of the primer had a greater influence on enzyme binding orientation than those near the 3′ end (Fig. 3b). To test this notion further, we constructed a new chimaeric primer 9D:10R with 5′-proximal DNA and 3′-proximal RNA, which had the same DNA/RNA ratio as the 10R:9D primer but with a different 5′-end backbone composition. As expected, the 9D:10R and 10R:9D primers supported opposite binding orientations of RT that closely resembled the orientational distributions of RT bound to pure DNA and RNA primers, respectively (Fig. 3a).

a, Selected FRET distributions of H-labelled RT bound to 5*-labelled substrates containing various 19-nt chimaeric RNA:DNA primers hybridized to a DNA template. FRET distributions of other xR:yD substrates are shown in Supplementary Fig. 7. b, The free-energy difference ΔG between the high-FRET and low-FRET orientations plotted as a function of RNA content for both xR:yD (red) and 9D:10R (blue) chimaeras. Error bars indicate s.e.m. (n = 3).
Binding orientation determines enzymatic activity of RT
The observation that RT bound to the DNA and RNA primers with opposite orientations suggests a hypothesis: that primer extension activity is determined by the binding orientation of the enzyme. To test this model, we probed the DNA polymerase activity of RT on the DNA (19D) and RNA (19R) primers as well as the chimaeric primers 9D:10R and 10R:9D, each annealed to a 50-nt DNA template (Fig. 4a and Supplementary Fig. 8). RT was capable of extending the 3′ end of both 19D and 9D:10R rapidly, with a rate comparable to previously reported steady-state extension rates of DNA primers34,35, even though the 9D:10R primer contained a ribonucleotide sugar backbone at its 3′ terminus. Polymerase activity was strongly inhibited for both 19R and 10R:9D. Furthermore, the rate of primer extension correlated with the fraction of time for which the RT enzyme bound in the polymerase-component orientation (Fig. 4b). These results indicate that the binding orientation is the strongest determinant of the primer-extension activity, whereas the content of the sugar-phosphate backbone contacting the active site of DNA polymerase is less important for synthesis activity. Our results also suggest a surprising allosteric effect in which contacts between the 5′ end of the primer and the RNase H primer grip regulate the DNA polymerase activity by determining the orientation of the enzyme on the substrate.

a, Primer extension activity of RT assayed on four selected primers (19D, 19R, 9D:10R and 10R:9D) annealed to a DNA template. The fraction of primers that had been extended by more than one base is plotted as a function of time for the four primers (coloured circles). The data were fitted to a single-exponential decay (grey lines) to deduce the primer extension-rate constants. b, Rate constants of primer extension (red) correlate with the fraction of time that the RT bound in the high-FRET orientation conducive to polymerization (blue).
Dynamic binding orientations of RT on PPT substrates
Although RNA primers do not generally support the initiation of DNA synthesis by RT, two copies of 15-nt RNA purine sequences, referred to as the PPTs, uniquely serve as primers for plus-strand DNA synthesis2,11,12,13,15. During infection, RT cleaves precisely at the 3′ terminus of the PPT, allowing DNA synthesis to be initiated at this position. The enzyme then removes the PPT primer by cleaving at its junction with the nascent DNA2,15. How RT interacts with the PPT to support the activities of both DNA polymerase and RNase H is still unknown. To address this question, we constructed three oligonucleotides encoding the PPT sequence to mimic different stages in plus-strand DNA synthesis. To simulate a PPT sequence that has not yet been cleaved at its 3′ terminus, we introduced a 2-nt RNA extension, creating the PPT:r2 RNA. Similarly, the PPT:d2 chimaera (containing a 2-nt DNA extension) was used to emulate a plus-strand primer from which DNA synthesis has already started. These primers were annealed to a 50-nt DNA template and assayed for RT binding (Fig. 5a–c). The FRET distribution of RT bound to the PPT:r2 primer–template complex was quantitatively similar to that observed for a non-specific RNA primer (compare Fig. 5a with Fig. 2d), suggesting that RT was predominantly bound in a cleavage orientation. By contrast, on the PPT and PPT:d2 substrates, RT spent a substantial portion of time in the high-FRET, polymerization-competent orientation (Fig. 5b, c). These data suggest that the priming activity of the PPT for plus-strand DNA synthesis originates from its specific ability among RNA sequences to direct RT binding in a polymerase-competent orientation.

a–c, FRET histograms of H-labelled RT bound to substrates containing 5*-labelled PPT:r2 (a), PPT (b) or PPT:d2 (c) primers annealed to DNA templates. The PPT sequence is shown in violet letters and the 2-nt RNA and DNA extensions are coloured orange and black, respectively. The DNA template is shown as a black arrow. d, FRET time traces of RT bound to PPT, PPT:d2 and PPT:dd2 substrates, showing spontaneous flipping transitions between the two binding orientations. e, FRET histograms of RT bound to PPT:dd2 substrates in the presence of 0, 10 μM and 1 mM dTTP. f, FRET histograms of RT bound to PPT:d2 substrates in the presence of 0, 10 and 100 μM nevirapine.
On the substrates that support both DNA-polymerase-competent and RNase-H-competent orientations, including the PPT, PPT:d2 and chimaeric RNA:DNA primers, RT exhibited spontaneous transitions between these two orientations, and flipping transitions were observed with different labelling schemes (Fig. 5d and Supplementary Fig. 9). The flipping transition did not seem to require the binding of multiple enzymes, because the flipping kinetics were independent of the RT concentration. The observation of flipping transitions within a single binding event was unexpected, considering the extensive network of contacts between the RT and its substrates19,22.
Small-molecule ligands regulate RT binding orientation
To explore the flipping mechanism, we investigated the effect of small molecules, including dNTP and non-nucleoside RT inhibitors (NNRTIs), on the equilibrium and rate constants of the flipping transitions by using the PPT:d2 primer or a modified primer containing a chain-terminating dideoxyribonucleotide (PPT:dd2). The addition of dTTP, the next cognate nucleotide for primer extension, stabilized the high-FRET, polymerase-competent orientation of RT (Fig. 5d, e). The stabilization magnitude increased with dTTP concentration over a physiologically relevant range (Fig. 5e). Kinetically, the addition of 1 mM dTTP decreased the rate constant of flipping from the high-FRET to the low-FRET orientation, khigh–low, 20-fold without substantially affecting the reverse rate, klow–high (Supplementary Fig. 10). By contrast, the addition of a mismatched nucleotide (dCTP) did not induce a similar effect (Supplementary Fig. 11).
NNRTIs are clinically approved anti-HIV drugs36 that bind to a hydrophobic pocket4 near the polymerase active site of RT to inhibit DNA synthesis allosterically37. We examined one such NNRTI, nevirapine, for its effects on the orientational dynamics of RT. Nevirapine seemed to have an opposite effect to that of cognate dNTP. The addition of nevirapine significantly destabilized the high-FRET, polymerase-competent orientation (Fig. 5d, f): the presence of the drug caused a 3.5-fold increase in the flipping rate from the high-FRET to the low-FRET orientation, khigh–low, without significantly altering the reverse rate, klow–high (Supplementary Fig. 10). A similar effect was observed for a different NNRTI, efavirenz. These results provide a structural basis for the previously observed specific inhibition of the PPT-initiated plus-strand DNA synthesis by NNRTIs, which occurs at a 40-fold lower concentration of NNRTI than that required for the inhibition of minus-strand DNA synthesis38.
Discussion
We have developed a single-molecule FRET assay to monitor the interactions between HIV RT and its nucleic-acid substrates in real time. These experiments directly revealed two opposite orientations with which the RT enzyme binds to DNA and RNA primers. The primary determinant of the enzyme orientation is the sugar backbone composition of the four or five nucleotides at each end of the primer, located within the polymerase and RNase H primer grip regions of the RT binding cleft. The primer-extension activity of RT is quantitatively correlated with the enzyme orientation, providing a structural basis for the primer-selection rule of RT.
Remarkably, the enzyme can bind to the special PPT RNA sequence, which directs the transition from minus-strand to plus-strand synthesis, in both orientations. Furthermore, the enzyme can flip spontaneously between the two states despite the extensive contact between RT and its nucleic-acid substrates. The flipping kinetics were altered by both cognate nucleotides and non-nucleoside RT inhibitors, but the two types of small molecule had opposite effects. Whereas the addition of cognate nucleotides caused a drastic decrease in the flipping rate from the polymerase-competent to the RNase H-competent orientation, the NNRTI substantially increased the same rate constant. NNRTIs and dNTPs have been shown to have opposite effects on the structural dynamics of the fingers and thumb subdomains20: whereas dNTPs bring these regions closer together to form a tighter clamp on the nucleic-acid substrate, NNRTIs cause further separation of the two subdomains. Our data therefore suggest a potential pathway for the flipping transition that requires relaxation of the ‘grip’ formed by the fingers and thumb subdomains around the nucleic-acid substrate. This spontaneous structural reorganization of the RT–substrate complex potentially allows the enzyme to rapidly explore multiple binding orientations that support distinct functions, thereby increasing replication efficacy.
Methods Summary
For single-molecule measurements of RT interactions with nucleic acids, the dye-labelled nucleic-acid substrates were immobilized on poly(ethylene glycol)-coated fused quartz slides through a biotin–streptavidin linkage. Binding of the dye-labelled RT molecules in solution to the immobilized substrates was monitored with the TIRF imaging geometry with alternating 532-nm and 635-nm excitations25. FRET histograms were constructed from binding events of hundreds of molecules. To calculate the difference in free energy between high-FRET and low-FRET states, these histograms were fitted to a double gaussian function and ΔG was determined from the ratio between the areas under the two gaussian peaks. Within each binding event, high-FRET and low-FRET substates were identified, and the lifetimes of the binding events and the substates were recorded. These lifetimes were combined with a simple kinetic model to derive the rate constants of transition between the substates as well as the rate of dissociation from each substate.
Online Methods
Preparation and labelling of RT and nucleic-acid substrates
The H-labelled RT was derived from a mutant enzyme in which a single-residue modification E478Q (ref. 30) was introduced to the RNase H domain to inhibit the RNase H activity. This modification was employed to prevent substrate cleavage during the single-molecule measurement. To engineer the H-labelled RT, native cysteine residues located at positions 38 and 280 were changed to serine, and a unique cysteine residue was introduced at the carboxy terminus of the p66 subunit to allow specific dye labelling through a thiol-maleimide reaction39. F-labelled RT was created from a similar mutant containing a unique cysteine residue at position 38 of the p66 subunit. Purified RT was incubated with Cy3-maleimide (GE Healthcare) and allowed to react for 60 min in 100 mM phosphate buffer (pH 7). The Cy3-labelled RT was then purified by dialysis for more than 48 h to remove the unreacted dye molecules. The p51 subunit was unlabelled.
Synthetic DNA (Qiagen Operon) and synthetic RNA (Dharmacon) oligonucleotides were purified by PAGE. All template strands (50 nt long) contained a biotin moiety attached to the 3′ end as well as an internal amino modifier (dT C6). The amine group was labelled with a monoreactive Cy5 in accordance with the manufacturer’s instructions (GE Healthcare) and the labelled template strands were HPLC-purified by reverse-phase chromatography on a C8 column (GE Healthcare). The primer strands (19–21-nt long) were annealed at 60 °C for 10 min to the template strands (in 10 mM Tris-HCl pH 8, 80 mM NaCl, 1 mM EDTA) at a roughly 15:1 ratio, cooled to less than 30 °C over about 1 h, and stored at -20 °C. Removal of unannealed primer stands was not necessary because they did not contain either dye molecules or biotin groups for surface immobilization.
Single-molecule FRET measurements of RT-substrate binding orientation
Quartz slides were cleaned with the use of argon plasma (Harrick Scientific), treated with 1% (w/v) Vectabond (Vector Laboratories) in acetone, rinsed, and then incubated with 20% (w/v) methoxy-PEG (Mr 5,000; Nektar Therapeutics) and 0.2% biotin-PEG (Mr 5,000; Nektar Therapeutics) in 0.1 M sodium bicarbonate (pH 8.4) for at least 3 h. Streptavidin (0.2 mg ml-1; Molecular Probes) and BSA (0.5 mg ml-1, New England Biolabs) in 10 mM Tris-HCl (pH 8), and 10 mM NaCl was applied to the slide before immobilization of the biotinylated primer–template complexes (50 pM).
Donor and acceptor fluorescence signals were collected on a prism-type TIRF microscope. The FRET donor Cy3 was excited by a 532-nm YAG laser (Crystal Laser) and direct excitation of Cy5 was performed by a 635-nm laser (Coherent). Emissions from donor and acceptor were separated with dichroic mirrors (Chroma Technology) and imaged onto the two halves of an Andor Ixon 887 back-illuminated electron-multiplying charge-coupled device. The FRET value is defined as IA/(IA + ID), where IA and ID are the fluorescence signals detected from the acceptor and donor channels, respectively, under 532-nm excitation.
During image acquisition, Cy3-labelled RT (6–24 nM) was added to the sample containing surface-immobilized primer–template complexes in an imaging buffer containing 40 mM NaCl, 50 mM Tris-HCl pH 8.0, 6 mM MgCl2, 0.1 mg ml-1 BSA, 10% w/v glucose and about 1.5 mM Trolox (Sigma Aldrich)40. An oxygen scavenger system (300 μg ml-1 glucose oxidase, 40 μg ml-1 catalase) was also added to the sample to decrease photobleaching. Positions of Cy5-labelled primer–template complexes were initially located by direct excitation of Cy5 with the 635-nm laser. FRET between Cy3 and Cy5 and fluorescence of Cy5 from direct excitation were then monitored by alternating the 532-nm and 635-nm excitations.
By increasing the intensity of the 532-nm excitation, photobleaching of Cy3 on the p66 subunit could be induced, shortening the duration of the observed fluorescence signal during a binding event. Under these conditions, Cy3 photobleaching was observed to occur in a single step, as manifested by the one-step decrease in the fluorescence signal to the background level, indicating that only one Cy3 dye was present on the enzyme–substrate complex. We therefore conclude that most of the binding events involved one p66 subunit. Because the RT concentrations used in the single-molecule imaging experiments (10–20 nM) were far below the equilibrium constant Kd for p66–p51 dimer formation (200–300 nM)41, an excess of unlabelled p51 was added (about 300 nM) to ensure that most p66 subunits formed dimers with p51. Because the p66–p51 heterodimers are more stable than the p66–p66 homodimers, the probability of potential p66–p66 dimer formation during the experiment was thus minimal under these conditions. Finally, because the p66 and p51 subunits alone showed much lower affinities than the p66–p51 dimers for the nucleic-acid substrates, we conclude that most of the binding events observed in the experiments involve a single p66–p51 dimer.
DNA polymerase activity measurements by single-molecule FRET
To confirm that RT retains DNA polymerase activity on surface-immobilized primer–template complexes, we performed an in situ primer extension assay by using single-molecule FRET. We designed a primer–template complex capable of reporting primer extension with FRET: the duplex region was labelled with Cy5, whereas Cy3 was placed at the 5′ end of the template, 10 nt from the 3′ end of the primer and 19 nt from the Cy5 (Supplementary Fig. 2a). Extension of the primer by the DNA polymerase activity of RT converted the single-stranded region of the template to double-stranded, stretching the template and lowering the FRET from about 0.8 to about 0.5 (Supplementary Fig. 2b). To monitor primer extension in real time, FRET time traces from the primer–template complexes in a single field were recorded while 100 nM unlabelled RT and 250 μM dNTPs were added. The in situ primer extension rate was determined by averaging all trajectories that showed a strong fluorescent signal and non-zero FRET (to avoid complexes with bleached dyes) and fitting the average trace with a single-exponential decay. This rate was compared with the results obtained from the unimmobilized primer–template complexes incubated with unlabelled or Cy3-labelled RT with the gel electrophoresis assay, as described below (Supplementary Fig. 2c).
DNA polymerase activity measurements by gel electrophoresis
To measure the DNA polymerase activity of Cy3-labelled RT in comparison with unlabelled RT, 100 nM Cy3-labelled or unlabelled RT was preincubated for 10–15 min in 150 μl of imaging buffer (described above) containing each dNTP at 250 μM. Extension of Cy5 end-labelled primer was initiated by the addition of 1.5 μl of 500 nM annealed primer–template complexes to the preincubated solution of RT and dNTP, and aliquots were removed at various time points, mixed in 90% v/v formamide, 1 × TBE buffer and 10 mM EDTA to quench the reaction, and heated to 90 °C for 1 min to denature the primer–template complexes. The products were then loaded on a precast 10% polyacrylamide gel (8 M urea; Bio-Rad). Intensities of fluorescent bands were measured with a Typhoon gel scanner (GE Healthcare). This condition was used to provide a better comparison with the single-molecule in situ data where RT and dNTP were added to the surface-immobilized substrate simultaneously. We note that the apparent primer extension rates measured in these experiments reflects the convolution of the binding rate of RT to the substrate and the true rate of elongation by RT, and thus should be slower than the true elongation rate as probed in the following experiments.
To measure the DNA polymerase activity of the RT on substrates containing the 19D, 19R, 10R:9D or 9D:10R primers, 3 μM unlabelled RT was incubated in the imaging buffer with 500 nM primer–template complexes. The 5′ end of each primer strand was labelled with 32P. Primer extension was initiated by the addition of 1 mM dNTP. The reaction was quenched at various time points by the addition of 500 mM EDTA and formamide. The reaction products were heated to 90 °C for 1 min then separated on an 8% polyacrylamide gel. Radiolabelled primers were imaged on a PhosphoImager cassette with a Typhoon gel scanner. The fraction of extended primers was quantified as a function of time. These data were fitted to single-exponential decays with an offset to deduce the primer extension rate constant for each substrate. For the 19R and 10R:9D primers, the fit was constrained to asymptote at a value of 0.8, which was the saturated fraction of extended primers for 19D and 9D:10R.
References
Goff, S. P. in Fields Virology Vol. 2 (ed. D. M. Knipe & P. M. Howley) 1871–1940 (Lippincott Williams & Wilkins, Philadelphia, 2001)
Champoux, J. J. in Reverse Transcriptase (ed. A. M. Skalka & S. P. Goff) 103–118 (Cold Spring Harbor Laboratory Press, New York, 1993)
Hostomsky, Z., Hostomska, Z., Fu, T. B. & Taylor, J. Reverse transcriptase of human immunodeficiency virus type 1: functionality of subunits of the heterodimer in DNA synthesis. J. Virol. 66, 3179–3182 (1992)
Kohlstaedt, L. A., Wang, J., Friedman, J. M., Rice, P. A. & Steitz, T. A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256, 1783–1790 (1992)
Baltimore, D. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226, 1209–1211 (1970)
Temin, A. M. & Mizutani, S. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226, 1211–1213 (1970)
Aiyar, A., Cobrinik, D., Ge, Z., Kung, H. J. & Leis, J. Interaction between retroviral U5 RNA and the TψC loop of the tRNATrp primer is required for efficient initiation of reverse transcription. J. Virol. 66, 2464–2472 (1992)
Leis, J. P., Berkower, I. & Hurwitz, J. RNA-dependent DNA polymerase activity of RNA tumor viruses. 5. Mechanism of action of ribonuclease H isolated from avian myeloblastosis virus and Escherichia coli . Proc. Natl Acad. Sci. USA 70, 466–470 (1973)
Hansen, J., Schulze, T., Mellert, W. & Moelling, K. Identification and characterization of HIV-specific RNase H by monoclonal antibody. EMBO J. 7, 239–243 (1988)
Tanese, N., Telesnitsky, A. & Goff, S. P. Abortive reverse transcription by mutants of Moloney murine leukaemia virus deficient in the reverse transcriptase-associated RNase H function. J. Virol. 65, 4387–4397 (1991)
Finston, W. I. & Champoux, J. J. RNA-primed initiation of moloney murine leukemia-virus plus strands by reverse-transcriptase in vitro . J. Virol. 51, 26–33 (1984)
Omer, C. A., Resnick, R. & Faras, A. J. Evidence for involvement of an RNA primer in initiation of strong-stop plus DNA-synthesis during reverse transcription in vitro . J. Virol. 50, 465–470 (1984)
Huber, H. E. & Richardson, C. C. Processing of the primer for plus strand DNA-synthesis by human immunodeficiency virus-1 reverse-transcriptase. J. Biol. Chem. 265, 10565–10573 (1990)
Schultz, S. J., Zhang, M. H., Kelleher, C. D. & Champoux, J. J. Analysis of plus-strand primer selection, removal, and reutilization by retroviral reverse transcriptases. J. Biol. Chem. 275, 32299–32309 (2000)
Rausch, J. W. & Le Grice, S. F. ‘Binding, bending and bonding’: polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. Int. J. Biochem. Cell Biol. 36, 1752–1766 (2004)
Gopalakrishnan, V., Peliska, J. A. & Benkovic, S. J. Human immunodeficiency virus type 1 reverse transcriptase: spatial and temporal relationship between the polymerase and RNase H activities. Proc. Natl Acad. Sci. USA 89, 10763–10767 (1992)
Wisniewski, M., Balakrishnan, M., Palaniappan, C., Fay, P. J. & Bambara, R. A. Unique progressive cleavage mechanism of HIV reverse transcriptase RNase H. Proc. Natl Acad. Sci. USA 97, 11978–11983 (2000)
Arnold, E. et al. Structure of HIV-1 reverse transcriptase/DNA complex at 7 Å resolution showing active site locations. Nature 357, 85–89 (1992)
Ding, J. et al. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol. 284, 1095–1111 (1998)
Huang, H., Chopra, R., Verdine, G. L. & Harrison, S. C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 (1998)
Sarafianos, S. G. et al. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 21, 6614–6624 (2002)
Sarafianos, S. G. et al. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 20, 1449–1461 (2001)
Stryer, L. & Haugland, R. P. Energy transfer: a spectroscopic ruler. Proc. Natl Acad. Sci. USA 58, 719–726 (1967)
Ha, T. et al. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl Acad. Sci. USA 93, 6264–6268 (1996)
Kapanidis, A. N. et al. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science 314, 1144–1147 (2006)
Stone, M. D. et al. Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature 446, 458–461 (2007)
Myong, S., Bruno, M. M., Pyle, A. M. & Ha, T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science 317, 513–516 (2007)
Rothwell, P. J. et al. Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc. Natl Acad. Sci. USA 100, 1655–1660 (2003)
Metzger, W., Hermann, T., Schatz, O., Le Grice, S. F. & Heumann, H. Hydroxyl radical footprint analysis of human immunodeficiency virus reverse transcriptase–templatėprimer complexes. Proc. Natl Acad. Sci. USA 90, 5909–5913 (1993)
Schatz, O., Cromme, F. V., Gruninger-Leitch, F. & Le Grice, S. F. Point mutations in conserved amino acid residues within the C-terminal domain of HIV-1 reverse transcriptase specifically repress RNase H function. FEBS Lett. 257, 311–314 (1989)
Nowotny, M., Gaidamakov, S. A., Crouch, R. J. & Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121, 1005–1016 (2005)
Nowotny, M. et al. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell 28, 264–276 (2007)
Julias, J. G., McWilliams, M. J., Sarafianos, S. G., Arnold, E. & Hughes, S. H. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo . Proc. Natl Acad. Sci. USA 99, 9515–9520 (2002)
Hsieh, J. C., Zinnen, S. & Modrich, P. Kinetic mechanism of the DNA-dependent DNA polymerase activity of human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 268, 24607–24613 (1993)
Kati, W. M., Johnson, K. A., Jerva, L. F. & Anderson, K. S. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 (1992)
El Safadi, Y., Vivet-Boudou, V. & Marquet, R. HIV-1 reverse transcriptase inhibitors. Appl. Microbiol. Biotechnol. 75, 723–737 (2007)
Spence, R. A., Kati, W. M., Anderson, K. S. & Johnson, K. A. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 267, 988–993 (1995)
Grobler, J. A. et al. HIV-1 reverse transcriptase plus-strand initiation exhibits preferential sensitivity to non-nucleoside reverse transcriptase inhibitors in vitro . J. Biol. Chem. 282, 8005–8010 (2007)
Rausch, J. W., Sathyanarayana, B. K., Bona, M. K. & Le Grice, S. F. Probing contacts between the ribonuclease H domain of HIV-1 reverse transcriptase and nucleic acid by site-specific photocross-linking. J. Biol. Chem. 275, 16015–16022 (2000)
Rasnik, I., McKinney, S. A. & Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods 3, 891–893 (2006)
Venezia, C. F., Howard, K. J., Ignatov, M. E., Holladay, L. A. & Barkley, M. D. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry 45, 2779–2789 (2006)
Acknowledgements
We thank J. Miller for providing initial samples of RT, and S. Liu for discussions. This work is supported in part by grants from the National Institutes of Health (GM 068518) and the Packard Foundation to X.Z., and from the Intramural Research Program of the Centre for Cancer Research, National Cancer Institute (to S.F.J.L.G.). X.Z. is a Howard Hughes Medical Institute investigator. E.A.A. is a Jane Coffin Childs postdoctoral fellow. Nevirapine and efavirenz were provided through the AIDS Research and Reference Reagent Program of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figures
The file contains Supplementary Figures 1-11 with Legends. (PDF 2884 kb)
Rights and permissions
About this article
Cite this article
Abbondanzieri, E., Bokinsky, G., Rausch, J. et al. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature 453, 184–189 (2008). https://doi.org/10.1038/nature06941
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature06941
This article is cited by
-
Biophysical approaches promote understanding of the viral replication cycle
Journal of the Korean Physical Society (2021)
-
Elucidating the in vivo interactome of HIV-1 RNA by hybridization capture and mass spectrometry
Scientific Reports (2017)
-
Analysis of HIV-1 intersubtype recombination breakpoints suggests region with high pairing probability may be a more fundamental factor than sequence similarity affecting HIV-1 recombination
Virology Journal (2016)
-
Genetic encoding of DNA nanostructures and their self-assembly in living bacteria
Nature Communications (2016)
-
Single-molecule imaging of non-equilibrium molecular ensembles on the millisecond timescale
Nature Methods (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
