
Abstract
Here we report the genome sequence of the honeybee Apis mellifera, a key model for social behaviour and essential to global ecology through pollination. Compared with other sequenced insect genomes, the A. mellifera genome has high A+T and CpG contents, lacks major transposon families, evolves more slowly, and is more similar to vertebrates for circadian rhythm, RNA interference and DNA methylation genes, among others. Furthermore, A. mellifera has fewer genes for innate immunity, detoxification enzymes, cuticle-forming proteins and gustatory receptors, more genes for odorant receptors, and novel genes for nectar and pollen utilization, consistent with its ecology and social organization. Compared to Drosophila, genes in early developmental pathways differ in Apis, whereas similarities exist for functions that differ markedly, such as sex determination, brain function and behaviour. Population genetics suggests a novel African origin for the species A. mellifera and insights into whether Africanized bees spread throughout the New World via hybridization or displacement.
Similar content being viewed by others
Main
The western honeybee, Apis mellifera, is a striking creature, one of relatively few species for which evolution culminated in advanced society1. In ‘eusocial’ insect colonies, populations are differentiated into queens that produce offspring and non-reproductive altruistic workers that gather and process food, care for young, build nests and defend colonies. Remarkably, these two castes, both highly derived relative to solitary insects, develop from the same genome.
Social evolution endowed honeybees with impressive traits2,3. Differentiation into queens and workers is through nutritionally based, hormone-mediated, programmes of gene expression4 yielding dramatic distinctions in morphology, physiology and behaviour. Queens, typically one per colony, have ten times the lifespan of workers, typically 1 to 2 yr5, lay up to 2,000 eggs per day, and store sperm for years without losing viability. Workers, numbering tens of thousands per colony, display sophisticated cognitive abilities, despite a brain containing only one million neurons6. This is five orders of magnitude less than the human brain and only four times greater than Drosophila, which has a far simpler behavioural repertoire. Workers learn to associate a flower’s colour, shape, scent, or location with a food reward7, increasing foraging efficiency. They communicate new food discoveries with ‘dance language’, originally deciphered by von Frisch8, the only non-primate symbolic language. Recent studies revealed that honeybees can learn abstract concepts such as ‘same’ and ‘different’9.
The infamous African ‘killer’ bees, Apis mellifera scutellata, the queens of which were introduced to Brazil in 195610, are known for intense stinging activity during nest defence, and pose human health problems. The African bees’ spread throughout the New World is a spectacular example of biological invasion. Although it was one of the first biological invasions to be studied with molecular tools11, our understanding of its genetic basis has been controversial.
This array of fascinating features, as well as amenability to molecular, genetic, neural, ecological and social manipulation12, led to selection of the honeybee for genome sequencing by the National Human Genome Research Institute, National Institutes of Health (NHGRI, NIH)13. The United States Department of Agriculture (USDA) also supported the project because of the paramount importance of pollination to human nutrition and the environment14. And, of course, humans and other animals have valued honey since prehistoric times.
Honeybees belong to the insect order Hymenoptera, which includes 100,000 species of sawflies, wasps, ants and bees. Hymenoptera exhibit haplodiploid sex determination, where males arise from unfertilized haploid eggs and females arise from fertilized diploid eggs. Haplodiploid-induced asymmetries in relatedness between offspring and sisters have long been thought to be involved in the evolution or maintenance of eusociality in the Hymenoptera15,16, but other life history traits also promote social evolution17, and there are divergent perspectives on this issue at the present time1,18. Haplodiploidy has distinct sex-determination mechanisms compared with other organisms because Hymenoptera lack sex chromosomes19.
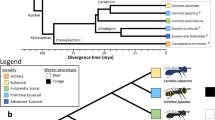
Hymenoptera is one of 11 orders of holometabolous (undergo a metamorphic moult) insects. All completed insect genome sequences have thus far been confined to Holometabola20,21,22,23,24,25,26; phylogenetic relationships of these and related arthropods are in Fig. 1. Honeybees diverged from Diptera and Lepidoptera 300 million years ago, whereas the last common ancestor with humans was 600 million years ago27. The genus Apis is an ancient lineage of bees that evolved in tropical Eurasia28 and migrated north and west, reaching Europe by the end of the Pleistocene epoch, 10,000 yr ago. The origin of A. mellifera has been suggested as Asia28, the Middle East29, or Africa2,30. From there, humans carried them worldwide because of their ability to make honey28.

Evolutionary relationships of Apis mellifera, other insects and related arthropods for which the genome sequence has been published (red), is in draft assembly form (blue), or is approved for sequencing (green), with approximate divergence times161,180. Recent work suggests that the Hymenoptera are basal to the Coleoptera in the Endopterygota (also known as Holometabola)76,152.
The A. mellifera genome has novel characteristics and provides fascinating insights into honeybee biology. Some main findings are:
• The A. mellifera genome is distinguished from other sequenced insect genomes by high A+T content, greater spatial heterogeneity of A+T content, high CpG content, and an absence of most major families of transposons.
• The honeybee genome evolved more slowly than that of the fruitfly and malaria mosquito.
• The A. mellifera genome shows greater similarities to vertebrate genomes than Drosophila and Anopheles genomes for genes involved in circadian rhythms, RNA interference (RNAi) and DNA methylation, among others.
• Apis mellifera has fewer genes than Drosophila and Anopheles for innate immunity, detoxification enzymes, cuticle-forming proteins and gustatory receptors, but more genes for odorant receptors, and novel genes for nectar and pollen utilization. This is consistent with honeybee ecology and social organization.
• Genes encoding the major royal jelly protein family—nine genes evolved from one ancient yellow gene—involved in queen and brood nursing, exemplify genes gaining new functions during the evolution of sociality.
• Novel microRNAs (miRNAs) were detected and shown to have caste- and stage-specific expression, suggesting a role in social diversification.
• Key elements in early developmental pathways differ between Apis and Drosophila, indicating that these evolved after the lineages separated.
• The honeybee shows similarities to Drosophila for functions that differ markedly, such as sex determination, brain function and behaviour.
• Population genetic analyses using new genome-based single-nucleotide polymorphisms (SNPs) support a hypothesis involving an African origin for the species A. mellifera and provide new insights into the spread of Africanized ‘killer’ bees. A. m. scutellata alleles have largely replaced those from one previously dominant subspecies, A. m. ligustica, whereas A. m. mellifera genotypes were essentially unchanged30.
Genome sequencing and assembly
The honeybee genome was sequenced using DNA from multiple drones derived from a single, slightly inbred queen (DH4 strain; Bee Weaver Apiaries, Inc.). 2.7 million whole-genome shotgun reads (Supplementary Tables 2 and 3) were assembled using the Atlas software31 and built into chromosomes using a microsatellite marker linkage map32,33. Analysis of initial assemblies showed that regions with high A+T composition were under-represented in libraries. Previously as much as 30% of the genome was in an (A+T)-rich shoulder in density gradients34. Thus, DNA was fractionated by CsCl-bisbenzimide density gradient centrifugation35 and additional shotgun libraries were generated from DNA with >70% A+T composition. Batches of 200,000 reads of (A+T)-rich DNA were generated and new genome assemblies were reassessed. After four such batches were added to the assembly, representing about 30% of the data, coverage of the (A+T)-rich regions had improved to 6-fold and the N50 (N50 is the contig size where 50% of the genome sequence is in contigs of size N50 or larger) of contigs had doubled to over 30 kilobases (kb) (Supplementary Fig 2 and 3), statistics adequate for gene predictions and analysis. In total, 1.8 gigabases (Gb) were assembled, ×7.5 coverage of the (clonable) 236 megabase (Mb) honeybee genome (Table 1, Supplementary Tables 2 and 3). Further details are available in Supplementary Information.
Several assemblies were produced (Supplementary Table 1) and are available on the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) website36, with statistics displayed in Table 1. Versions 1.x were initial assemblies deficient in (A+T)-rich regions. Version2 (January 2005) was the first to contain full (A+T)-rich read enrichment. The genome size and coverage of this assembly did not change for later assemblies. Version2 lacks highly repeated sequences, which has little impact on gene predictions, and used the 2005 version of the microsatellite linkage map (from M. Solignac) to build chromosomes. In version3, highly repeated sequences were added, which increased the N50 (see below) for contigs by 15% and for scaffolds by 6%. With the current (March 2006) version4 assembly, the most recent genetic map (AmelMap333) was used to place sequence on chromosomes, increasing the amount of mapped sequence by 10%.
Assemblies were tested against honeybee data sets (expressed sequence tags (ESTs), complementary DNAs, microsatellite markers and sequenced bacterial artificial chromosome (BAC) clones) for quality and completeness (Supplementary Table 3). The quality of version3 and version4 was tested by alignment of full-length cDNAs (55 total). All alignments showed correct order and orientation of exons. Although a limited set of cDNAs, it nevertheless seems that misassembly of genic regions is rare. In version4, 99% of 2,032 markers and 98% of 3,136 ESTs were represented, providing evidence for completeness. When aligned to BACs that had been independently sequenced, 23 out of 27 BACs showed >94% coverage, whereas four were >80% but were complex owing to repeated sequences. The estimated clonable genome size is thus taken as 231 Mb/98% = 236 Mb. Flow cytometry estimates with nuclei from brains of 45 workers and 12 drones gave a haploid genome size of 262 ± 1 Mb. The difference between drones (264 ± 2) and workers (261 ± 1) was not significant (P ≫ 0.05). Compared to the assembly, we estimate unassembled or unclonable (for example, paracentromeric (A+T)-rich, see below) sequences at 26 Mb.
Sets of gene predictions were produced using version2 (see below and Supplementary Information). Analyses proceeded using versions 2, 3 and 4, depending on the particular characteristic under study and timing of the assembly.
Genetic and physical maps and chromosome organization
Development of linkage maps was facilitated by a high recombination rate, 19 cM Mb-1, several-fold greater than in any other multicellular eukaryotes32,37,38. The linkage map AmelMap3 has more than 2,000 microsatellite markers. The average distance between markers is 2.1 cM; all intervals are shorter than 10 cM. The high density of this map suggests that little information is missing in the assembly. Scaffolds were organized along chromosomes according to this map. Sixty-four per cent (151 Mb) of scaffolds contain at least two markers with non-null distances and could be oriented on chromosomes, whereas another 15% (35 Mb) contained one marker and could be placed but not oriented. Thus, 79% (186 Mb) of the genome is placed on chromosomes.
The honeybee karyotype (Fig. 2) is based on measurements of morphologically distinct features of chromosomes in 74 well-spread, 4,6-diamidino-2-phenylindole (DAPI)-stained haploid chromosomes prepared from testis of drone pupae of the sequenced strain. Measurements of centromere positions, overall chromosome length, position of ribosomal organizer regions, and position and extent of (A+T) sequence-rich (DAPI-positive bands) are in Supplementary Table 4. The total length of the 16-chromosome haploid complement at meiotic pre-metaphase is 30 μm. Of this, 36% (11 μm) is (A+T)-rich, DAPI-positive sequence surrounding each centromere. The chromosomes are numbered according to the genetic map, which orders them roughly by length, ranging in size from 3.5 μm for metacentric chromosome 1 to 1.2 μm for chromosome 16. All chromosomes (plus nuclear organizing regions containing rDNA repeats on chromosomes 6 and 12) have been identified by BAC fluorescence in situ hybridization (FISH) using one or more BACs containing microsatellites used in the genetic mapping effort. This karyotype largely agrees with previous efforts39; however, it is not possible to pair unequivocally the several similar-sized chromosomes in these two karyotypes.

The ideogram (in blue) shows average chromosome lengths, positions and sizes of DAPI-positive (heterochromatin) bands. The percentage of heterochromatin reflects the time of appearance of heterochromatic bands (100% observed in all preparations; lower percentages seen only in early prophase spreads). Lines to the right of chromosomes represent BACs shown by FISH to bind in relative order and positions predicted from the genetic and physical maps. Binding sites of rDNA probes (distal short arms of chromosomes 6 and 12) are shown in red. The karyotype (below the ideogram) is based on the rightmost spread.
Manual superscaffolding of chromosomes 13, 14, 15 and 16
Additional evaluation of completeness of version4 was performed using ‘manual superscaffolding’ to bridge gaps between mapped scaffolds on the four smallest chromosomes. The requirement for two unconflicted mate pairs to link contigs was relaxed and all additional sources of evidence were used. These included overlaps of contigs that were not merged because of haplotype divergence, single-mate-pair links between contigs, extensions of contigs using trimmed parts of reads yielding novel overlaps, resolution of conflicting mate pairs, and cDNAs and confident gene models bridging inter-scaffold gaps. Attempts were made to cross the 22 remaining inter-superscaffold gaps by polymerase chain reaction (PCR), with five being successful. This effort reduced chromosomes 13–16 (17% of the mapped genome) from 21, 25, 42 and 21 mapped scaffolds to 4, 5, 6 and 5 superscaffolds, respectively, and incorporated 121 unmapped scaffolds totalling 1.8 Mb for an increase in length of 5.5% for these chromosomes (superscaffolds are available from BeeBase).
The resultant superscaffolds extend from the mapped location of the centromere—although centromeric sequences were not discerned—to the TTAGG telomeric repeats of the distal telomeres (see below). Comparison with the genetic map suggests that the remaining inter-superscaffold gaps are not extensive. Only two misassemblies of 146-kb and 65-kb sections of scaffolds, as well as several minor misassemblies of 2–8-kb contigs, were discovered in this 17% of the mapped assembly. This manual effort provides additional support for the near completeness of the assembly for the euchromatic regions of the genome.
Genome organization
Although the A. mellifera genome is not the first sequenced insect genome, a number of characteristics of genome organization distinguish it. Described below, the honeybee provides new diversity in genome structure.
Sequence characteristics
Animal genomes are a mosaic of G+C-content domains, with homogeneous G+C composition within domains, but widely variable G+C composition between domains. In all animals studied, including the honeybee, the distribution of G+C-content domain lengths follows a power law distribution (for example, ref. 40). The G+C-content domains in the honeybee genome, as in other genomes, do not have a characteristic length; rather, there is an abundance of short segments and only a small number of long ones. Comparison of G+C-content domain lengths in various genomes (Supplementary Fig. 4) shows that honeybee domains are shorter than in two dipterans, which are shorter than chicken and human. The honeybee genome is more (A+T)-rich than other sequenced insect genomes (67% A+T in honeybee, compared with 58% in Drosophila melanogaster and 56% in Anopheles gambiae, Fig. 3a).

a, G+C-content domain length versus G+C percentage in A. mellifera (green), A. gambiae (blue) and D. melanogaster (red). The dashed line at 20% G+C content indicates the large number of low-G+C domains in A. mellifera. b, Gene length (top) and transcript length (bottom) versus G+C percentage of G+C content domains in which genes are embedded. Gene length was computed as the genomic distance from start to stop codon of the longest splice variant of each gene. Transcript length was computed as the distance between start and stop codon on the transcript sequence.
Consistent with an (A+T)-rich genome, honeybee genes occur more frequently in (A+T)-rich domains compared with other species (Fig. 3b and Supplementary Fig. 5). The mean G+C content of domains in which honeybee genes are located is 29%, compared with 47% for A. gambiae and 44% for D. melanogaster. Furthermore, genes are not distributed evenly throughout the genome, but show a tendency to appear in (A+T)-rich regions of the honeybee genome (C. Elsik et al., personal communication).
Gene length in the honeybee shows a striking relation to G+C content compared with other insects (Fig. 3b). Gene length (exons plus introns) increases with G+C content in honeybee (R2 = 0.135, P < 2.2 × 10-16), but decreases slightly with G+C content (R2 = 0.009, P = 1.1 × 10-10) in A. gambiae, and is not significantly related to G+C content (R2 = 0.0002, P = 0.079) in D. melanogaster. However, transcript length (exons only) has a significant (P < 2.2 × 10-16) but weak positive correlation with G+C content in all three insects (A. mellifera R2 = 0.067, A. gambiae R2 = 0.037, D. melanogaster R2 = 0.033). These relationships in honeybee indicate that total intron length increases with G+C content, in contrast with vertebrates in which intron length decreases with G+C content41. The relationship between gene length and G+C content is unlikely to be the result of annotation bias. First, this analysis included only genes that were flanked on both sides by other genes on the same scaffold, avoiding genes with exons on different scaffolds. Second, honeybee gene models were a consensus of five gene sets (see below), and were associated with probabilistic confidence scores. The relationship between gene length and G+C content did not change when gene models with less than 90% probability were removed from the analysis. Thus the honeybee genome is unusual among insects and opposite of vertebrate genomes in having long genes in (G+C)-rich regions.
Among dinucleotides, CpG is over-represented (1.67-fold) compared with the expectation from mononucleotide frequencies. Such an excess contrasts with other eukaryotic genomes where CpG is under-represented or at most close to the expected value (the highest CpG content so far is 1.15-fold in the genome of Cyanidioschyzon merolae42). Genomes where CpGs are the target of cytosine methylases (such as plants or vertebrates) show a CpG deficit (Supplementary Table 5). The high CpG content in the honeybee is intriguing because honeybee is the first protostome shown to possess a full complement of functionally active vertebrate-like DNA methytransferases, including an active CpG methytransferase (see below). Methylated cytosines are known to be hypermutable43, and Me-C→T mutations are expected, driving base composition towards A+T richness. However, the impact of cytosine methylation on mono- and dinucleotide composition in the honeybee is not clear.
Telomeres
The 15 acrocentric honeybee chromosomes have a distal telomere on their long arm and a proximal telomere on their short arm, whereas the large metacentric chromosome 1 is presumably a centromeric fusion of two acrocentric chromosomes with two distal telomeres and loss of the proximal telomeres39 (Fig. 2 and Supplementary Table 4). We built all 17 distal telomeres44. Twelve were already present at the ends of terminally mapped scaffolds, whereas the remainder were assembled by manually superscaffolding outwards from the terminally mapped scaffolds. After 1–7 kb of unique sequence beyond the last gene on each Apis chromosome, each distal telomere has a 3–4-kb subtelomeric region, showing 70–92% sequence identity between all 17 telomeres. This is followed by the expected TTAGG45 or variant telomeric repeats of at least several kilobases. The canonical organization of these distal telomeres (Fig. 4) makes them the simplest and most consistently constituted telomeres known in insects. In contrast, Bombyx mori telomeres are complicated by the insertion of numerous non-long terminal repeat (LTR) retrotransposons46, and dipteran telomeres are unconventional in having no TTAGG repeats: Drosophila telomeres consist entirely of multiple non-LTR retrotransposon inserts of the het-A and TART families47,48,49, and the telomeres of A. gambiae50 and Chironomus midges51 consist of many complex tandem repeats, maintained by recombination.

Organization of centromeric-proximal telomeres on the short arm of the 15 acrocentric chromosomes in Apis is hypothetical based on FISH studies. (The blue checked area represents the 176-bp tandem AluI repeats (see text).) Bombyx46 and Drosophila47 telomeres are based on one or two telomeres in each species. Regions of non-LTR retrotransposons are indicated. For the Drosophila subtelomeric region (telomere-associated sequence repeats, TAS), shading indicates the presence of short (50–130 bp) repeat sequence blocks. Telomeres of A. gambiae50 and Chironomus midges51 consist of complex tandem repeats, as indicated.
We have been unable to build the 15 proximal telomeres, which also appear by FISH to have the TTAGG repeats45, much as we have been unable to assemble the 16 centromeres, although their approximate map locations are known52. They are composed of highly repetitive tandem sequences of two major kinds: the 176-bp AluI repeat at proximal telomeres and the 547-bp AvaI repeat in centromeres39. These two repeats constitute 2% and 1% of the genome respectively, yet in the current assembly only a few short unmapped scaffolds contain them, whereas the vast majority remain in the unassembled-repeat-read data set. We conclude that the distal and proximal telomeres differ in their subtelomeric region sequences, even if both have terminal TTAGG telomere repeats. This subtelomeric difference might be important for the 15 acrocentric chromosomes, where in the Rabl configuration after mitosis the distal telomeres are sequestered on the nuclear envelope at one pole of the nucleus opposite the telophase centromeric cluster, as well as in the chromosome bouquet formation during meiotic prophase53. To allow this Rabl configuration the proximal telomeres must be distinct, most likely in their subtelomeric regions, and hence remain with the centromeres. The apparent absence of AluI repeats from the single metacentric centromere region is consistent with this interpretation39.
Transposable and retrotransposable elements
Almost all transposons identified are members of the mariner family54,55,56, widespread in arthropods and other animals, with relatives in plants, protists and bacteria57 (Table 2 and Supplementary Information). They range in age from the relatively young AmMar1 (refs 58, 59) with many nearly intact copies, to the ancient AmMar5 and AmMar6 with only a few highly degraded copies remaining. Other common types of transposons are largely absent from the honeybee genome (Supplementary Information).
There is little evidence for active retrotransposable elements, but the genome once harboured many retrotransposons. There are remnants of several LTR retroviral-like retrotransposons; for example, 15 partial and highly degraded copies of a copia-family sequence, 6 partial sequences encoding matches to the BEL12 element of A. gambiae60, and 3 highly degraded copies of a DIRS retrotransposon61. There are also remnants of a further possible 11 LTR- and 7 non-LTR retrotransposons from a number of clades identified in Drosophila62. Separate manual assembly of nuclear rDNA units revealed that honeybees do contain active non-LTR retrotransposons of the R2 family, although the R1 lineage seems to be absent63. There are, however, at least five short degraded copies of an R1-like element most similar to the R1Bm element of B. mori64, so R1 elements were also once present. This relative lack of retrotransposons is a surprising finding in light of high diversity, copy number and activity of both LTR and non-LTR retrotransposons in other insect genomes. Possibly retrotransposons are too disruptive to a genome that is completely exposed to selection in haploid drones every generation, with the rDNA being a relative ‘safe haven’, although retrotransposons are known from other haplodiploid Hymenoptera65,66,67,68.
It is apparent that this is an unusual genome in having few transposons and retrotransposons, at least in assembled sequences, almost all of which are members of the mariner family constituting 1% of the assembled genome. The vast majority of these mariner copies and the degraded retrotransposons are in short unmapped scaffolds, suggesting that they might reside in poorly assembled pericentromeric regions that might constitute heterochromatin, and that there might be additional copies within the unassembled centromeric regions, just as there are R2 elements in unassembled rDNA repeats. Genomic screens for highly repetitive sequences identified only three major classes that represent about 3% of the genome, mapping to the centromere and telomere by chromosome in situ analyses39,45. This 3% portion of the genome does not assemble well and could harbour undetected transposons.
Gene content and the proteome
The gene list
Five gene lists, each produced using the version2 assembly, were combined to produce a master gene list: the Official Gene Set (OGS). The component gene sets were from NCBI, Ensembl, Softberry (Fgenesh), an evolutionarily conserved core set and a set based on Drosophila orthologues (Supplementary Information). The gene sets were merged using GLEAN, which uses latent class analysis to estimate accuracy and error rates for each source of gene evidence, and then uses these estimates to construct a consensus prediction based on patterns of agreement or disagreement observed between each evidence source69. The five input gene sets and merged GLEAN set were compared to each other using FASTA70 to assess accuracy and completeness in representing 395 protein sequences that had been manually curated but not used to make gene predictions (a ‘gold standard’ collection of genes for evaluating different gene lists). The GLEAN set of 10,157 genes was superior by several measures (Supplementary Table 6) and was deemed to be the OGS. This set is considered as the list of genes that are based on experimental evidence. A second list of genes was constructed from gene predictions that were not strictly based on experimental evidence, the Official ab initio Gene Set (OAIGS), and comprised 15,500 Fgenesh gene models that did not overlap genes in the OGS. The annotation consortium manually annotated over 3,000 gene models using standard operating procedures developed by community members and BCM-HGSC. Most annotated gene models were from the OGS because most OAIGS models were not valid (see below). Global comparisons to other organisms relied on the OGS, whereas BLAST searches to identify honeybee orthologues of known genes (and families) used both the OGS and OAIGS.
Validation with whole-genome tiling array data
A complete transcription profile of the honeybee genome was analysed at high resolution using tiling arrays (M. P. Samanta et al., personal communication). Seven million 36-mer oligonucleotide probes were selected to represent the A. mellifera genome, including all intergenic regions, then queried with pooled honeybee messenger RNA from multiple tissues and life stages (Supplementary Information). An established statistical technique was applied to determine whether an annotated gene was transcribed71 (Supplementary Information). Sixty-seven per cent of genes in the OGS were expressed; in contrast, only 5% of genes in the OAIGS were expressed. Similarly, less than 1% (4 out of 456) of OAIGS genes from 15 long (G+C)-rich chromosomal segments lacking OGS genes showed expression in another experiment. These results provide independent empirical support for the GLEAN-derived OGS. Transcriptional signals were also observed in 2,774 intergenic regions. Some of these novel transcripts are non-coding RNAs.
Validation via annotation of chromosome 15 and 16 superscaffolds
As another assessment of the quality of the OGS, manual annotation was performed. Because this is a time-consuming process, the two smallest chromosomes, chromosomes 15 and 16, were selected and their entire superscaffolds were manually annotated by carefully inspecting gene models from all gene prediction sets after BLAST comparison to known protein and EST/mRNA sequences (Supplementary Information). For chromosomes 15 and 16 respectively there were 720 and 337 gene models created, with 5 and 7 tRNAs, 5 and 14 pseudogenes with multiple frameshifts, and 71 and 62 splice variants; 188 and 116 gene models of the OGS were significantly corrected by adding/removing exons, adjusting splice sites and merging/splitting transcripts. Only 48 and 23 new protein-coding gene models were added to the OGS (less than 1%), including 40 and 15 that were previously supported by only Fgenesh ab initio gene models, consistent with the tiling array analysis. Only 56 and 21 transcripts on these two chromosomes (7%) were problematic due to assembly gaps or indels (insertions and/or deletions) affecting open reading frames, consistent with the view that the draft assembly is high quality in predicted genic regions. A putative function was assigned to 639 and 254 protein-coding genes for chromosomes 15 and 16, respectively.
MicroRNAs
Two computational surveys of the genome for miRNA-like sequences identified 65 candidate miRNAs (Supplementary Table 7), including orthologues of confirmed miRNAs in other organisms, and novel miRNA candidates comprised of micro-conserved elements. Seventeen candidates, including eight novel miRNAs, were selected for validation by RT–PCR.
Some putative miRNAs, including novel candidates, exhibited caste-, stage- and/or tissue-specific expression profiles, ranking among the top 10% of all tiling array signals (D. Weaver et al., personal communication). For instance, two novel miRNAs (C5599F, C689F) were more strongly expressed in queen abdomen than in worker, are among the strongest tiling array signals, and are in human and Drosophila genomes. C689F is also one of the eight most abundant miRNA-like transcripts in pupae. By contrast, novel miRNA C5560 displays differential developmental stage specificity and is more strongly expressed in worker pupae than queen, but is more abundant in all tissues and castes than any other putative miRNA tested. Thus, miRNAs may have a function in developmental regulation of social organization, and some new miRNA candidates may have diverse roles in regulating gene expression in other organisms.
Gene order in insects
As in vertebrates72, gene order in insects is under limited selection. Less than 7% of single-copy orthologues retain gene order in three-way genome comparisons of Apis, Drosophila and Anopheles. This fraction is about 10% in an Apis–Drosophila comparison, considerably lower than chicken–human (over 85%)72 although both genome pairs diverged approximately 300 million years ago. This discrepancy can be attributed to higher rates of genome evolution in insects (see ref. 73).
Beyond local gene arrangement, chromosome-level synteny can be established for species as divergent as Drosophila and Anopheles74,75 as both have five major chromosomal elements. However, only a few correspondences can be established between the 16 Apis chromosomes and Drosophila and Anopheles chromosomes. There are significantly more orthologues shared between Drosophila chromosomal arm 3R and Apis groups 5 and 15 as well as Drosophila chromosomal arm 2L and Apis group 4, pointing to a common origin76, whereas no other chromosome regions between Apis and Drosophila show statistically significant enrichments of shared orthologues.
Orthology and rate of honeybee evolution
Single-copy orthologues conserved among many species are well suited for measuring differences in genome evolutionary rates. All genes were classified based on their homology to genes in other completely sequenced organisms, using only three vertebrates (human (Homo sapiens), chicken (Gallus gallus) and fish (Tetraodon nigroviridis)) in order to obtain a relatively balanced data set in terms of species divergence (Fig. 5 and Supplementary Tables 8–10). To characterize the conserved core of honeybee genes and estimate evolutionary rates from it, we identified single-copy orthologues likely to be present in all metazoans. In the OGS, approximately 30% fall into this category (for exact definitions, see Fig. 5). For the remainder (70%) of the predicted honeybee genes, orthologous relationships are more complex, involving many-to-many orthologues and nonuniformly occurring (patchy) orthologues, or not detectable. These genes are indicative of functional differences at various levels. For example, there are 1,052 honeybee genes with orthologues that are unique to the three insects considered (that is, they probably encode insect-specific phenotypes). This set compares to 3,816 human genes with orthologues only in the three vertebrates (see striped boxes in Fig. 5), suggesting a more complex gene pool coding for vertebrate-specific features.

At the extremes, genes might be part of the metazoan core proteome (darkest band, bottom) or unique to a species with currently no counterpart in other organisms (white band, top). The striped boxes indicate insect- and vertebrate-specific genes and show that there are far fewer in insects. ‘1:1:1’ indicates universal single-copy genes, but absence or duplication in a single genome is tolerated as we cannot exclude incomplete genomes or very recent duplications. This explains uneven numbers between species of these very conserved metazoan core genes. ‘X:X:X’ indicates any other orthologous group (miss in one species allowed), with X meaning one or more orthologues per species. ‘Patchy’ indicates other orthologues that are present in at least one insect and one vertebrate genome. ‘Homology’ indicates partial homology detected with E < 10-6 but no orthology classified.
Comparison of the 2,404 single-copy orthologues present in exactly one copy in each of the insects and in human revealed that the mean sequence identity between honeybee and human is considerably higher than that of fly and human (47.5% versus 44.5%, with t-test significance of 10-11, see Fig. 6 and Supplementary Fig. 6) and also higher than between mosquito and human (46.6%). This indicates a considerably faster evolutionary rate in the fly lineage and points to the honeybee as the slowest evolving insect sequenced so far. Two other independent measures confirmed this observation, as various genome-based rate measures tend to give similar results73. Patchy orthologous groups, defined as having at least one member lost in both insects and vertebrates (Fig. 5), contain non-essential genes that are relatively easy to lose during evolution. The high fraction of retained genes within this category in the honeybee shows that it lost fewer genes than either one of the Diptera species (Fig. 6; the higher number of losses in mosquito compared to fly might be due to lower quality of mosquito annotation). When comparing the fraction of conserved intron positions (Fig. 6) between insect genes and vertebrate orthologues, a massive loss of introns in Diptera becomes apparent whereas the honeybee has kept almost 80% of the identifiable ancient introns since divergence from vertebrate ancestors. The higher number of introns in the honeybee (see Supplementary Table 11 and Supplementary Fig. 7 for more data on six genomes) also supports the conclusion that the honeybee is slower diverging and less derived than the other insects—fly and mosquito. This observation is reflected in other orthologue categories in Fig. 5. For example, the honeybee has fewer many-to-many orthologues than fly and mosquito, indicating slower evolution (but this might be partially due to a more stringent honeybee gene prediction). Taken together, analysis of honeybee orthologues is in accordance with a recent finding that early metazoans had many, intron-rich genes77 that have been subsequently lost and/or simplified (in terms of intron numbers) in fast evolving lineages such as insects, wherein the honeybee seems to be less reductive than the two Diptera species—fly and mosquito.

Comparison of single-copy orthologues in honeybee, fly and mosquito versus human in terms of: average protein identity; retained fraction of ‘patchy’ orthologous groups, as defined in Fig. 5; and fraction of retained ancient introns (those that are found in at least one of the vertebrate orthologues; positional conservation was counted within sliding windows of ±10 bases to allow for intron sliding). The standard error of the mean is about 0.3% and is shown by the error bars.
There are several clear indications of honeybee-specific duplications implying unique functions. A striking example is farnesyl pyrophosphate synthase (FBgn0025373), known to be involved in lipid metabolism in fly, which occurs as a single copy in all other sequenced insects whereas there are seven copies in the honeybee. When strictly requiring single-copy orthologues in all five other metazoan genomes analysed, we find 60 genes that have been duplicated only in the honeybee (Supplementary Table 12). The fixation of these duplicates of otherwise single-copy orthologues is rare and has been shown to be associated with the emergence of species-specific functions78. Some of these may be relevant to the solitary versus social lifestyle differences between these insect groups.
In addition to the presence and absence of genes and gene families, the expansion and reduction of the latter indicates, at a global level, change of functionality. By ranking the most extreme differences between honeybee and fly in terms of domain family occurrences (Supplementary Table 13), the major contributors to phenotypic change become visible (Fig. 7). The most extreme cases in honeybee include expanded families of odorant receptors and the major royal jelly proteins, which are important in caste differentiation, as discussed below.

The top five most prominent expansions and contractions of InterPro-defined protein or domain families in A. mellifera, D. melanogaster and H. sapiens. The families are ordered by the chi-squared test significance of the family size difference with respect to the predicted number of genes, 10,157 and 13,450, respectively. Absent families are marked. 7TM, seven transmembrane.
Apis genes shared with deuterostomes but missing from Drosophila
The D. melanogaster genome sequence revealed the absence of many genes and pathways that are present in other animals such as mammals and nematodes, and sometimes yeast20. Comparison with the Anopheles genome showed that some of these genes are present in other insects and hence must have been lost from the Drosophila lineage75. Automated comparisons with the OGS suggest 762 proteins that honeybees share with at least one deuterostome but which appear to be missing from Drosophila. A similar number are missing from Anopheles, but about 300 proteins in Apis are missing from both of these dipterans. They were presumably lost early in dipteran evolution or sometime between the divergence of Hymenoptera from the lineage that led to Diptera. These candidate losses of otherwise widespread and conserved animal genes from the Drosophila and more broadly fly lineages provide opportunities for many studies. Two of these have been published: the Mahya protein expressed in the mushroom bodies and other brain regions implicated in memory and learning79, and pteropsin, a non-visual vertebrate-like opsin expressed deep in honeybee brains and possibly involved in linking the circadian clock to daylight80,81. Other examples, such as telomerase, Dnmt1, Dnmt3 and SID-1 are discussed below.
Functional categories
Using the orthologue set described above, the honeybee genome was compared with other genomes, in particular with the well-annotated, finished Drosophila and human genomes to identify protein and domain families for which gene number was different. An important theme in this analysis was relating such differences to the social lifestyle of the honeybee. Table 3 gives some examples of obvious changes whereas Table 4 focuses specifically on neurobiology and behaviour, as discussed below. There also are many examples in which there are subtle changes in the honeybee genome, and some examples including venom components, heat shock proteins, functions for nectar and pollen utilization, and antioxidant systems are presented in the Supplementary Information.
Development
Signalling pathways
A small number of highly conserved cell signalling pathways—Wnt, hedgehog (Hh), transforming growth factor-β (TGF-β), receptor tyrosine kinase (RTK), Notch, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and nuclear hormone—are responsible for most developmental cell–cell interactions in metazoans82. In honeybees, like most metazoans, the genes encoding components of these pathways are conserved; however, the components of some more unusual cell signalling systems, those that specify the early axes in Drosophila, are missing from the honeybee genome83. Of the genes that specify terminal embryo fate, trunk and torso are absent from the honeybee, implying that terminal patterning occurs through a different pathway from that of Drosophila and Tribolium castaneum. The gene gurken, a component of the Drosophila dorso-ventral signalling system, is also missing from the honeybee genome.
This absence of early-acting, axis-specifying genes extends to those that are not involved in cell signalling. Both bicoid, an anterior specifying gene, and oskar are missing from the honeybee, Tribolium and Bombyx mori genomes84. Orthodenticle and hunchback genes, which replace bicoid in Tribolium, are present in the honeybee. Oskar acts as a pole plasm anchor in Drosophila85, and pole plasm is absent from Tribolium86, Bombyx87 and the honeybee88.
The absence of some of the earliest acting factors in these pathways is consistent with the hypotheses of refs 89 and 90, which postulate that the initial steps in a developmental cascade are likely to have evolved most recently. It will be important to study how each signalling/patterning system works in the honeybee without these key factors, and whether their activity has been replaced by other, unknown, rapidly evolving genes.
Homeobox genes
Ninety-six homeobox domains were found in 74 genes, either alone or in combination with PAX, POU, LIM and other domains (Supplementary Table 14), similar to Drosophila20. The HOX cluster genes are in one cluster on chromosome 16 and the genes are transcribed from the same strand83. This is one of the rare syntenic blocks found in the honeybee and fly genomes.
Sex determination, lack of dosage compensation and male meiosis
The honeybee shows genomic similarities to Drosophila for sex determination despite marked differences in this process (Fig. 8)83. Males receive a random half of the mother’s genome under a haplodiploid mode of reproduction; there are no gender-specific chromosomes. Sex in the honeybee is determined by the allelic composition of a single locus called complementary sex determiner (csd, Fig. 8). Despite the lack of sex chromosomes, honeybees have putative orthologues of some of the genes in Drosophila—run, sc and dpn91—although these are not involved in sex determination in the honeybee and their function is unknown. Most genes downstream in the pathway have orthologues but there is no honeybee emc homologue. In the fly cascade, tra, which controls somatic sex differentiation, has no orthologue in the honeybee, but the honeybee’s initial sex determination signal csd is thought to be a functional equivalent of tra19. Orthologues of dsx and ix are found in the honeybee genome. Honeybee dsx is sex-specifically spliced19, consistent with a conserved sex-determining function in both flies and honeybees. These divergent pathways functionally converge at the dsx gene.

Sex in the honeybee is determined by the allelic composition of a single gene, the complementary sex determiner (csd)19. Eggs develop into males when csd is hemizygous (haploid) or homozygous, or females when csd is heterozygous. Honeybees lack sex chromosomes and X-specific dosage compensation. Sex-specific information is transferred in both species from diverged initial signals to the final gene, dsx, via switch genes that are active (on) in the females, but inactive (off) in the males. Most Drosophila pathway genes are present in the honeybee genome despite the marked differences (see text).
Despite the lack of X-specific dosage compensation, the honeybee has orthologues of mle, mof, msl-3 and Trl, which control dosage compensation in Drosophila. It is possible that these genes have additional functions in the honeybee. No potential orthologues of msl-1, msl-2, roX1 or roX2 were identified in the honeybee genome.
Because male honeybees are haploid they lack meiosis. Gene Ontology (GO) analysis identified seven genes in D. melanogaster that are involved in male but not female meiosis. Only three of these genes have orthologues in the honeybee (bol, crl, topi). Seven genes specifically involved in female meiosis in Drosophila subjected to the same analysis showed only four with orthologues in the honeybee, possibly indicating that several genes involved in the process of meiosis are fast evolving. Most genes of the fly’s sex determination pathway are conserved in the honeybee despite the marked differences in sexual regulation. It is of interest to understand whether the evolutionary conservation of the genes results from functions other than sex determination that are shared among the honeybee and the fly. This will further support the notion that the early-acting factors of the pathway have been recruited more recently to the sex-determining function and dosage compensation89,90.
Caste, reproduction and ageing
Brood feeding
Royal jelly, produced by exocrine glands in the head of adult worker bees, is an important component of the food used in cooperative brood care, and a key factor in caste differentiation3. The genes encoding the major royal jelly proteins (MRJP) provide one of the best examples of a gene family gaining new functions during the evolution of sociality92. The MRJP family in honeybees is encoded by nine genes arranged in a 65-kb tandem array. The MRJP protein family seems to have evolved from a single progenitor gene that encodes a member of the ancient yellow protein family. Five genes encoding yellow-family proteins flank the genomic region containing the genes encoding MRJPs93.
Caste development
Previous studies identified ESTs representing genes differentially regulated during caste differentiation94,95,96,97. Improved genome-based annotation of these ESTs and GO analysis of their Drosophila orthologues has led to new insights into caste determination.
Genes associated with metabolic regulation are prominent in the EST sets related to caste, corroborating earlier suggestions that changes in metabolism are particularly important in caste determination94,95,96,97. For example, queens and workers show different gene expression patterns for oxidoreductases (overexpressed in queen larvae) and hydrolases (overexpressed in worker larvae). In addition, genes overexpressed during worker development are better defined in terms of GO categories than are genes that are overexpressed during queen development (without GO attributes are 0 out of 17 worker overexpressed genes, in contrast to 9 out of 34 genes overexpressed in queens). Even considering the limits in transferring GO terms from Drosophila to honeybee, this finding suggests that the evolution of sterility in the worker caste will ultimately be explainable in terms of known molecular processes. The results also suggest that it may be possible to gain insights from gene expression analyses of caste development in a highly eusocial species into basic questions in socioevolution, namely, what was gained by splitting functions normally performed by a solitary hymenopteran female into two or more castes, and how was this split integrated into post-embryonic differentiation to generate truly alternative phenotypes98.
Insulin/insulin-like growth factor signalling
Insulin/insulin-like growth factor signalling pathways are well-conserved integrative pathways regulating ageing, energy metabolism, fertility and other important biological processes. In Caenorhabditis elegans, 37 genes encoding putative insulin-like ligands have been identified, whereas 7 have been identified in D. melanogaster. However, both C. elegans and D. melanogaster have a single insulin/IGF-1 receptor orthologue.
There are some notable differences between honeybees and C. elegans/D. melanogaster for components of insulin/insulin-like growth factor signalling pathways. Honeybees have only two genes encoding insulin-like peptides (ligands) (AmILP-1 and AmILP-2) and two putative insulin/IGF-1 receptors (AmInR-1 and AmInR-2). Honeybees have four orthologues of C. elegans daf-16 (regulator of longevity) compared with one in Drosophila (dFOXO). These differences in ligand/receptor stoichiometry suggest that honeybees may have evolved a different regulation of this complex pathway. Perhaps these differences also relate to their striking reversal in the traditional relationship between fertility and lifespan. In most organisms, high fertility is achieved at the expense of longevity, whereas in honeybees and other social insects, this relationship is converted into a positive one: queens are both highly fecund and long lived. Recently discovered connections between insulin signalling and the classical insect hormones99,100 might provide a link between differential expression of metabolism-related genes in caste development, local patterning in morphogenetic fields and endocrine signals driving metamorphosis. Results from the genome project will increase the effectiveness of the honeybee as a model to examine how the insulin/insulin-like growth factor signalling pathway could have been modified to extend lifespan without negatively affecting reproductive capabilities.
Cuticular and peritrophic membrane proteins
Cuticle consists primarily of chitin and its associated proteins. The most abundant class of cuticular proteins has an extended R&R consensus (pfam00379) that binds to chitin101. The 28 R&R proteins identified in Apis are less than one-third of the number of genes for putative cuticular proteins with that domain found in Drosophila or Anopheles. One possibility is that a less complex cuticle structure allowed by the protected hive environment accounts for the reduced number of genes. This speculation is supported by a comparison of another chitinous structure, the peritrophic membrane of the midgut that has proteins with chitin binding domains (CBD)102. The Apis genome has nine peritrophic membrane proteins with CBDs, a number comparable to those in other insect genomes; some have multiple CBDs.
Telomerase
Organismal ageing is also frequently attributed to a decline in telomerase activity. The presence of the canonical insect telomeric TTAGG repeat at the ends of both kinds of telomeres (distal and proximal) implies that, unlike dipterans, honeybees have telomerase. Using the human telomerase reverse transcriptase (TERT) sequence as a query sequence, we identified a candidate gene with 23% amino acid identity to human TERT44. The whole-genome tiling array (above) and quantitative RT–PCR analysis (M. Corona, H. M. Robertson and G. E. Robinson, personal communication) confirm the expression of telomerase in honeybees. Identification of the honeybee distal telomeres and telomerase will allow study of the possible involvement of telomere length and telomerase activity in the extreme ageing differences of worker, drone and queen honeybees.
Brain and behaviour
Honeybees display a rich behavioural repertoire and have long been recognized as a model system for the study of social interactions. A set of candidate honeybee genes for behaviours representing diverse signalling pathways has already been identified by a number of laboratories12. Here we explore differences in the diversity of various pathways and gene families previously implicated in brain function and behaviour, and how these might relate to honeybee behaviour (Table 4).
Ion channels, neurotransmitters and other signalling molecules
Similar to the genome of Drosophila, the A. mellifera genome encodes a conserved set of pore-forming voltage-gated ion channels, but is missing most of the auxiliary subunits found in vertebrates. In contrast to the ∼50 two-pore (TWIK) potassium channels present in C. elegans, the A. mellifera genome encodes only ten. A similar contraction of channel number occurs in the degenerin/amiloride-sensitive sodium channel family, where A. mellifera has only 8 genes compared to 24 in Drosophila. The classes and numbers of ligand-gated ion channels are largely similar between Drosophila and A. mellifera (Supplementary Fig. 8), with the interesting exceptions that the A. mellifera genome encodes three N-methyl-d-aspartate (NMDA) receptor subtypes, instead of two, and five glutamate excitatory transporters, instead of two. The honeybee also has one extra nicotinic acetylcholine receptor (nAcR) subunit103. A reduction of genes also holds true for regulatory and catalytic protein kinase A subunits. Moreover, one gene comprises the CREB/CREM family of transcription factors in the honeybee and Drosophila, whereas three genes were found in vertebrates104.
Neuropeptides modulate the behaviour and affect the activity of almost every neuronal circuit. Thirty-six brain peptide genes in Apis encode prohormones that are processed into an estimated 200 neuropeptides105, a number similar to that reported from Drosophila and Anopheles106,107,108. However, nine unique neuropeptide genes that do not fit within known neuropeptide families have been found in the honeybee genome. In addition to the neuropeptide genes themselves, 37 neuropeptide and protein hormone G-protein-coupled receptors (GPCRs) have been annotated (48 in Drosophila). Furthermore, 19 biogenic amine receptors have been found (21 in Drosophila), bringing the total number of honeybee neurohormone GPCRs to 56. The probable ligands for 39 of them have been identified109.
Apis, like Drosophila, lacks brain-derived neurotrophic factor (BDNF) signalling machinery, which is well conserved in vertebrates. However, the honeybee has all the components of the agrin synapse formation pathway, several of which, including agrin itself, are missing in flies. The core synaptic vesicle trafficking machinery is largely conserved between the currently sequenced invertebrate genomes, although the honeybee synaptotagmin family is more similar to mice and humans than Drosophila or Anopheles.
Chemoreceptors
In the honeybee genome there is a remarkable expansion of the insect odorant receptor family relative to D. melanogaster (62 odorant receptors from 60 genes)110,111,112 and A. gambiae (79 odorant receptors)113. A total of 170 odorant receptor genes were manually annotated, of which 7 are pseudogenes114 (this is twice the number of seven transmembrane odorant receptors in Fig. 7 because the automated annotations concatenated many tandemly arrayed genes). These constitute five honeybee-specific subfamily lineages in an insect odorant receptor family tree, and one of these lineages is hugely expanded with 157 genes encoding 15–99% amino acid identity. One-hundred and forty-two of these odorant receptor genes are in 14 tandem arrays of two or more genes distributed throughout the genome, including one of 60 genes, reflecting likely expansion by unequal crossing over. This huge odorant receptor family expansion presumably mediates the honeybee’s remarkable range of odorant capabilities, including perception of several pheromone blends, subtle kin recognition signals, and diverse floral odours. It is notable that this large number of odorant receptor genes corresponds well with the estimated number of 160–170 glomeruli in the honeybee antennal lobe115, consistent with the central model of insect olfaction, which holds that each olfactory receptor neuron expresses one112 or at most two116 odorant receptors. All olfactory receptor neurons expressing the same odorant receptors then converge on a single glomerulus per antennal lobe, allowing for an odour map of patterns of olfactory receptor neuron stimulation115.
The odorant receptor family is but one highly expanded lineage of a superfamily of several distantly related lineages of chemoreceptors111, most of which are implicated in gustatory function and therefore are called gustatory receptors117, although some might have olfactory functions116. In contrast to the D. melanogaster repertoire of 68 gustatory receptors encoded by 60 genes111 and the A. gambiae repertoire of 76 gustatory receptors encoded by 52 genes113, the honeybee gustatory receptor repertoire consists of just 10 unclustered gustatory receptors representing 7 divergent lineages114. The honeybee might have lost one or two ancient gustatory receptor lineages, but the difference is largely the result of differential expansion of subfamilies in the two flies and lack of any gustatory receptor subfamily expansion in the honeybee. The limited gustatory receptor repertoire perhaps reflects the provisioning of larval bees by adults and their reduced need to avoid toxic chemicals in their food—the relationship between honeybee and flower is mutualistic. Honeybees also antennate each other and other objects, and are perhaps using some odorant receptors as gustatory receptors.
Odorant binding proteins are a third major component of the insect chemosensory system, with the potential to influence chemosensation118. The honeybee genome encodes 21 odorant binding proteins, less than half the number of either dipteran, with 51 in Drosophila and 70 in Anopheles119. If odorant binding proteins function in a combinatorial fashion with the receptors, this relatively limited repertoire might restrict the range of honeybee olfaction; however, the roles of odorant binding proteins beyond transport of hydrophobic molecules in insect chemosensation remain unclear and many are not expressed in chemosensory organs.
Circadian rhythms
The honeybee orthologues of the clock proteins cryptochrome (Cry), timeless (Tim), clock (Clk) and cycle (Cyc), which are mostly associated with the ‘negative feedback’ loop of the clock, are more similar to mammals than Drosophila80. For example, the honeybee genome does not contain orthologues to dCry (Drosophila-type Cry) and timeless1 (tim1), genes that are essential for clock function in the central pacemaker of Drosophila, but does have orthologues to genes encoding the mammalian-type paralogues mCry (mammalian-type Cry) and timeout (Tim2), which are thought to have different clock function120. The temporal brain expression pattern of AmCyc, AmClk, AmCry and AmTim2 is more similar to mammals than to Drosophila, suggesting that they behave like their mammalian orthologues. Additional analysis suggests greater Apis–Drosophila similarities in the ‘positive feedback loop’ of the clock. The honeybee genome encodes highly conserved orthologues to Vrille (Vri) and PAR domain protein 1 (Pdp1), two basic zipper transcription factors that are implicated in the regulation of Clk expression in Drosophila, with the highest similarity (>94% identity, 100% similarity) in the DNA binding domain. On the other hand, the honeybee genome does not encode true orthologues to the orphan nuclear receptors Rev-Erb (α and β; nuclear receptor subfamily 1, group D members 1 and 2; NR1D1 and NR1D2, respectively) and ROR (α, β, γ; nuclear receptor subfamily 1, group F, members 1–3; NR1F1–3, respectively), which are thought to orchestrate the expression of Bmal1 (the vertebrate orthologue of cycle) in the mammalian clock121. Although the honeybee genome encodes related nuclear receptors (GB11364-PA, related to Ecdysone-induced protein 75B, a NR1D3 protein, and GB10650-PA, similar to Hormone receptor like 45, a NR1F4 protein), we found PAR family consensus elements (putative binding sites for Vri and Pdp1) but no RORE response elements (putative binding sites for ROR and Rev-Erb proteins) in the 3 kb upstream of the start codon of either AmClk or AmCyc. Phylogenetic analyses show that the basal animal lineage had both the mammalian and Drosophila types of Cry and Tim80 (Supplementary Fig. 9). Thus, Drosophila diverged by specializing on using one set of orthologues and by losing mCry; honeybees converged with mammals by losing these orthologues and specializing on the other set. These analyses of the honeybee genome sequence uncover previously unknown diversity in animal clocks, challenge the distinction commonly made between the clocks of insects and vertebrates, and raise critical questions concerning the evolution and function of clock genes.
Immunity and disease resistance
Honeybees live in highly crowded nests, providing favourable demographic conditions for infectious diseases. Honeybee pathogens are well known122 and include viral, bacterial, fungal and protist pathogens, along with other parasites123. Protection from infectious disease includes social defences such as grooming and other hygienic behaviours, individual chambers for raising young, and a workforce that defends the nest against many potential vectors of disease. Individual honeybees are also defended by morphological barriers and immune defences.
Curiously, given the predicted disease pressures in honeybee colonies, the honeybee genome encodes fewer proteins implicated in insect immune pathways when compared to other insect genomes. Although the Toll, Imd and JAK/STAT pathways seem to be intact, paralogue counts for gene families implicated in these pathways are reduced by two-thirds124. This reduction spans every step in the immune response from pathogen recognition to immune effectors, and implies a reduced flexibility in the abilities of honeybees to recognize and resist pathogens. The results suggest that honeybees use novel immune pathways, are poorly defended against pathogens at the individual level, and/or have immune systems that are narrowly focused on a relatively small group of coevolved pathogens.
Gene-expression and RNAi studies have begun to elucidate roles for several candidate immune genes in response to microbial infection125,126. Coupling these studies with efforts to understand pathogen gene expression127,128 and invasion mechanisms129, and with the long-standing search for the impacts of genotypic and environmental variance on disease resistance73,75, will cement the honeybee as an essential model for the study of immunity and disease in social insects.
Anti-xenobiotic defence mechanisms
Honeybees are vulnerable to insecticides and have suffered major population losses in some regions of the world. Contact pesticides affect the worker bees whereas residual pesticides accumulate in lipophilic substances such as wax or pollen lipids130 and impact on the developing brood and queen fecundity. Wax acts as a pesticide sink131 and pesticide residues incorporated into wax may migrate to honey132. Sub-lethal effects of pesticides affect honeybee initial learning and conditioned odour responses, traits directly linked to foraging.
It seems that the size of the major detoxifying gene families is smaller in the honeybee, making the species unusually sensitive to certain pesticides133,134,135,136. Compared with Anopheles and Drosophila137, the honeybee has 30–50% fewer genes encoding the carboxylesterase (CCE), cytochrome P450 (P450) and glutathione S-transferase (GST) enzymes that are principally responsible for the metabolism of pesticides and in which the great majority of metabolic resistance mutations have been found in other species of invertebrates138. The greatest difference is seen in the GSTs, the family most strongly associated with detoxifying functions. Two clades of GSTs containing all known insecticide-resistance-related GSTs in other insects, and comprising over 20 members in D. melanogaster and A. gambiae, consist of a single member in the honeybee. Similarly, the honeybee genome has just half the number of P450 genes and contains less than 20% of genes of the CYP4 clade (21 in Drosophila, 4 in the honeybee), which is strongly associated with pyrethroid resistance in other species.
Conversely, there is usually the same number of genes, and in some cases significantly more, in clades within the CCE, P450 and GST families that are not involved in detoxification. The marked variation in relative abundance argues strongly against explanations for changes in gene number in honeybee based on genome-wide factors like its haplodiploid genetic system and the associated exposure to selection in the haploid caste.
Gene regulation
DNA methylation
DNA methylation systems are well characterized in vertebrates139. By contrast, methylation in D. melanogaster and other invertebrates remains controversial140. Evidence for DNA methylation has been demonstrated in several different orders of insects, but these results are interpreted cautiously because no catalytically active deoxycytosine methyltransferase has yet been identified and characterized in any invertebrate. The honeybee genome contains genes that encode orthologues of all vertebrate proteins required for DNA methylation140. In addition to Dnmt2 (also found in Diptera), three CpG-specific DNMT family genes were identified: two Dnmt1 genes and one Dnmt3a/b gene. A single putative methyl-DNA-binding-domain-containing gene with two splicing variants was also identified. The DNMT genes are expressed and are active in vitro. Moreover, the honeybee methylation system is functional in vivo, as shown by genomic 5-methyl deoxycytosine in honeybee DNA and several specific CpG methylated endogenous gene sequences140.
RNA interference
The honeybee genome harbours only a single homologue of sid-1, a gene essential for systemic RNAi141, whereas multiple copies of this gene occur in the moth and draft Tribolium castaneum genomes84. The honeybee SID-1 homologue clusters with one of three beetle and two of three moth SID homologues (Fig. 9), as well as two vertebrate paralogues, one of which was lost in fish but preserved in birds and mammals.

The maximum likelihood phylogram (Phylip) is based on the alignments of the conserved carboxy-terminal transmembrane domain141 of SID-1 proteins from insects, vertebrates, nematodes and other eukaryotes. Sequences were aligned with Clustalw. The sequences are identified by species name and, where multiple genes exist in a genome, by a further identifier (see Supplementary Information). A long basal branch from Dictyostelium to the other sequences is truncated (thick bar).
This disparity in SID-1-like gene number may relate to honeybee RNAi efficacy. RNAi in honeybees was first demonstrated by embryo injection of double-stranded RNAs142, and later experiments showed that, as in other invertebrates, injection of double-stranded RNA can reduce target gene expression away from the site of injection143. However, the molecular details of honeybee gene silencing have not been clearly elucidated144, and little is known about the role of SID-1 proteins in insect systemic RNAi.
The honeybee genome also encodes many other proteins similar to the core RNAi machinery of other species, including two Dicer (Dcr) enzymes, a Drosha homologue and other RISC components, like argonaute 1 and 2, as well as a full suite of double-stranded RNA binding proteins—R2D2, Pasha and Loquacious—all of which have yet to be confirmed as components of the RNAi pathway in honeybees.
Nuclear receptor transcription factors
The set of 22 nuclear receptors encoded by the Apis genome is a nearly perfect match to the Drosophila set, a strong reminder of the centrality of these transcription factors in the regulation of insect embryonic development and metamorphosis145. The single novelty uncovered in Apis nuclear receptors is subtle: the presence in Apis of a third gene homologous to vertebrate photoreceptor-cell-specific nuclear receptor (PNR), represented in Drosophila by DHR51 and DHR83 (ref. 146). If, as predicted on the basis of studies of human PNR mutations, Apis PNR-like is involved in the differentiation of the visual system147,148, this additional gene may reflect a key aspect of the honeybee’s behavioural ecology: reliance on vision for navigation. This finding is consistent with an apparent overall increase (relative to Drosophila) in the number of genes putatively involved in photoreception81.
cis-Regulation of behavioural development
Adult worker honeybees typically shift from working in the hive to foraging for nectar and pollen outside the hive when they are 2–3 weeks of age, but the age at which this transition is made depends on the needs of the colony, which are communicated among honeybees via pheromones12. To begin to study the cis-regulatory code associated with this form of social regulation, the genome sequence was scanned149 for regulatory motifs in the promoter regions of genes expressed in the brain that are related to socially regulated behaviour development and identified via microarray analysis150,151.
Results of statistical analyses (see Supplementary Information) indicate that the transcriptional regulatory pathway for social behaviour in honeybees shares several commonalities with the pathway for development in Drosophila. Gene expression was significantly associated with binding sites for the transcription factors Hairy, GAGA, Adf1, CF1, Snail and Dri. Promoter sequence patterns predicted brain expression for as many as 71% of certain types of behaviour-related genes, even though less than 15% of all transcription factors known from Drosophila were studied. Social regulation of gene expression is a potent influence on behaviour in animals and humans12, and these results will help to elucidate underlying mechanisms.
Bee phylogeny and population genetics
Information from the honeybee genome is providing new insights into the origins of honeybees and their spread throughout the world. Ordinal relationships of the higher insects have been re-examined using concatenated sequences from 185 (ref. 152) and 1,150 (ref. 76) protein-coding genes. These new phylogenies support the hypothesis that the order Hymenoptera (ants, bees and wasps) is derived from an early branch in the holometabolous insects, which challenges the current view that the lineage leading to Hymenoptera diverged after the evolution of beetles (Coleoptera) (Fig. 1). Phylogenetic analysis of all bee families using sequences derived from the Honey Bee Genome Project supports a hypothesis that the Apidae branched off earlier in the phylogeny of bees than previously recognized153. This, combined with the fact that the oldest fossil bee (Cretotrigona prisca) is a corbiculate apid, supports the view that honeybees and their relatives are the oldest lineage of eusocial bees. Other eusocial bee lineages (Halictidae and Allodapini) evolved eusociality later than honeybees154.
The honeybee genome sequence was used as the basis for the development of a large set of SNPs. These have been used to generate a view of the relationships among A. mellifera subspecies of unprecedented detail. In the following sections we summarize the SNP resource, present an initial view of the relationships among A. mellifera subspecies, and summarize key findings30 on historical patterns of migration, differentiation and introgression of honeybees in both the Old and New World to help define the origins of domesticated honeybees and their resultant worldwide diaspora. The SNP set will also be essential for marker-assisted and positional cloning of genes that underlie important behavioural, social and economically relevant traits.
SNP resources
Candidate SNPs were identified from two sources: (1) alignment of 2,483 genome traces from Africanized honeybees to the assembled genome sequence (European-derived); and (2) alignment of ∼75,000 ESTs derived from genetically variable European honeybees. Using a bayesian method155, 3,594 and 1,950 base-substitution SNPs from each source (respectively) showed a high probability (P > 0.99) of being true polymorphisms. An initial honeybee SNP panel was generated from 1,536 putative SNPs, of which 1,136 were validated (see Supplementary Information). These SNPs were genotyped in 328 A. mellifera (from native and introduced populations) and 13 individuals from three related species: Apis cerana, Apis dorsata and Apis florea. Details of these methods are presented in Supplementary Information. Results are summarized in the following section and described in more detail elsewhere30.
Biogeography and phylogeny of native and Africanized honeybees
Analyses of SNP genotypes in ten subspecies (Fig. 10) revealed four non-overlapping clusters of A. mellifera subspecies analogous with four evolutionary lineages (called M, C, O and A) defined by morphometric characters28,156 and consistent with mitochondrial DNA analysis157 (where C and O lineages, as defined here and in ref. 28, correspond to the ‘C’ mitochondrial type). Surprisingly, within A. mellifera, north and west European honeybees (M) were more similar to African (A) than to the geographically proximal east European honeybees (C) (Fig. 10). Phylogenetic analysis using outgroup genotypes from A. cerana, A. dorsata and A. florea suggested that extant A. mellifera subspecies originated in Africa30, consistent with speculation of an origin in tropical or subtropical Africa by ref. 2 but contrary to current hypotheses of an origin for A. mellifera in western Asia156 (based primarily on the occurrence of approximately ten allopatric species in eastern Asia). Taken together, these data support a hypothesis involving an African origin for A. mellifera and at least two separate migrations into Eurasia: a migration into Europe via the Iberian Peninsula, which has since expanded into central Europe and Russia (M group), and one (or more) migrations into Asia and east Europe south of the Alps (O and C groups, respectively).

Neighbour-joining tree using Nei genetic distance181. Ten geographical subspecies (N = 9–21 individuals each) can be partitioned into four regional groups. Branches separating regional groups are supported by 100% bootstrap.
Africanization in the New World has involved the near-replacement of the ‘European’ honeybee by descendents of A. m. scutellata. Analysis of SNP genotypes in New World bees30 revealed several key findings. First, pre-existing ‘European’ populations showed evidence of extensive admixture between C, M and O groups (consistent with known introductions from at least nine subspecies158). Second, although African alleles were dominant in populations after Africanization, all Africanized individuals showed evidence of introgression between A. m. scutellata and pre-existing populations. Third, replacement of European- by African-derived alleles was origin-dependent: pre-existing C group alleles were replaced but M group alleles were not. The explanation for the latter result is unclear, but may result from the close relationship between A and M (compared to A and C; Fig. 10) or from historical and local patterns of introductions.
Conclusions
After Mendel completed his work with peas, he turned to experimenting with honeybees, to extend his work to animals (http://www.zephyrus.co.uk/gregormendel.html)159. He produced a hybrid strain (so vicious they were destroyed) but failed to reproduce the clear picture of heredity because of difficulties in controlling the mating behaviour of queens159. The honeybee genome similarly proved a challenge for sequencing with biases in clone libraries. With special remedies we produced a draft sequence that is high quality by a number of metrics. The overall statistics for completeness (genome size, coverage of markers, ESTs and cDNAs) indicate that over 96% of these important elements are in the sequence. Similarly, assessments of quality indicated few misassemblies, mainly associated with repeated sequences, as expected for a draft sequence. We expect that regions remain with lower sequencing redundancy but these regions are not missing altogether and their sequences are included in homology searches and can be recognized. Other expected limitations are accurate placement of repeated sequences and heterochromatic regions, which are problematic even in sequences taken to the highest finished quality.
In addition to providing sequence, a genome project also produces a gene list, generated here by novel merging of five efforts69. The OGS consisted of a little more than 10,000 genes, lower than other insects (D. melanogaster, 13,600 genes20; A. gambiae, 14,000 genes23; B. mori, 18,500 genes26,160). Genome tiling array and manual annotation efforts increase the OGS by a few per cent, but it remains smaller by 15% or more compared with other sequenced insects. Because the sequence does not appear to be missing such an amount of the genome, we believe that the gene count is underestimated. Consistent with this are results from whole-genome tiling array experiments that detected signals in thousands of regions currently described as intergenic.
We suspect two reasons for the current low gene number. First, limited EST and cDNA data for Apis reduced gene predictions. Second, the large evolutionary distance of Apis from other sequenced genomes restricted use of orthology in predicting genes and may have introduced a bias in the OGS. When genes that are known in other organisms were not found in the OGS or the OAIGS, orthologues themselves were used to search the honeybee genome, and genes found were added to the OGS. But shorter genes, rapidly diverging genes, and other special cases may not be readily detected by this approach.
Members of honeybee gene families were less likely to be missed because other family members showed sufficient sequence similarity to be useful in searches. Supporting this is the observation of a number of expanded gene families; for example, the major royal jelly protein and odorant receptor families. We predict that the gene count for the honeybee will increase in the future as more data and analyses are applied.
The honeybee genome’s high A+T content, absence of transposons and slower rate of evolution, will be better understood as more insect genomes are sequenced161. The paradoxes of high CpG content despite the presence of cytosine methylases, fewer genes for innate immunity despite the high pathogen and parasite loads associated with social life, or the presence of Drosophila orthologues for many genes in the sex determination pathway despite the honeybee’s lack of sex chromosomes, promise to establish the honeybee as a new model for several fundamental processes of life.
A number of new resources have been produced to enhance biological discovery. The discovery of genes related to RNAi (for example, a SID-1-like protein) should enhance the use of this technique in the honeybee. It is difficult to select and propagate mutants in the haplodiploid, polyandrous, open-air-mating honeybee, making it difficult to apply traditional forward genetic tools. RNAi can now be used for clarifying gene function and altering protein expression for beneficial effects on honeybee behaviour or physiology. Equally important, the genome facilitates characterization of core molecular features of RNAi, potentially clarifying inconsistencies observed in silencing some honeybee gene targets. The discovery of novel miRNAs and cis-regulatory elements associated with social behaviour provides a foundation to begin to understand social regulation of gene expression. Another new resource is an extensive SNP set, which already has generated new insights into honeybee phylogeography and invasion biology, and will prove invaluable for positional cloning of genes in quantitative trait loci for a variety of traits such as defensive behaviour or foraging behaviour162.
How will the honeybee genome sequence enable a mechanistic understanding of social organization, communication and the ability to shape the local environment? The evolution of sociality requires changes to every system in the organism, not only to invent new functions but also to tune old ones to new purposes. We expected to find a rich set of genetic features underpinning honeybee sociality, and here again, the genome project has not disappointed.
One intriguing trend is a smaller size of some gene families relative to the other sequenced insect genomes, possibly reflecting a selective elimination of genes whose functions have become superfluous in the now highly specialized life history and self-managed environment of the honeybee. Larger gene family sizes are, however, also observed. New genes are not created de novo, but result from duplication and diversification. The initial analysis of the honeybee genome presented here shows 60 such duplications that are not present in other genomes. These, and others like them awaiting discovery, are candidates for honeybee-specific functions. The major royal jelly proteins provide a good example of protein family expansion and social evolution93.
However, achieving a comprehensive understanding of social life in molecular terms will require extensive analyses of the honeybee as well as other social and non-social species. A genome might be a blueprint for some aspects of biology, but most mysteries of sociality appear to be encoded subtly in the genome, at least based on our study of honeybee and Drosophila, as well as recent analyses of human and chimpanzee163. Although much remains to be done, with the genome in hand, and the associated methodologies it enables, prospects are bright for elucidating the molecular and genetic bases of many complex traits associated with honeybee sociality.
Methods
Detailed methods are described in Supplementary Information. Sources for resources generated by this project are listed here.
Genome assemblies
Genome assemblies are available from the BCM-HGSC ftp site under the directory (ftp://ftp.hgsc.bcm.tmc.edu/pub/data/Amellifera/fasta/) (see Supplementary Table 17). The files available for each assembly differ, but in general there are directories for contigs (the sets of contigs with fasta quality and agp files for each linkage group), for linearized scaffolds (sequences for each linkage group where the gaps between contigs have been filled with Ns), for bin0 (non-overlapping reads) and repeat reads. The file descriptions and assembly statistics for each version are described in a file named README.txt.
The version 4.0 assembly directory also contains small contigs (less than 1 kb) that were omitted from the assembly as well as haplotype contigs (overlapping contigs identified as representing the second haplotype in the sequenced DNA). The individual accessions at the NCBI are: version 4.0 (scaffolds CM000054–CM000069, CH876891–CH878241); version 3.0 (contigs AADG05000001–AADG05018946); version 2.0 (contigs AADG04000001–AADG04016028; scaffolds CM000054–CM000069, CH402995–CH404444); version 1.2 assembly (contigs AADG03000001–AADG03022771; scaffolds CM000054–CM000069, CH236967–CH239577); version 1.0 (contigs AADG02000001–AADG02030074); version 1.0 (contigs AADG01000001–AADG01015795). The version 2.0 assembly is displayed in the NCBI Map Viewer.
Genome browsers
Genome browsers are available for viewing the genome assemblies at BeeBase (http://racerx00.tamu.edu/bee_resources.html), NCBI (http://www.ncbi.nlm.nih.gov) and UCSC (http://genome.ucsc.edu). The assembly versions are listed in Table 1 and Supplementary Table 1. The feature annotations that are available differ from site to site.
Chromosome superscaffolds
Chromosome superscaffolds for chromosomes 13, 14, 15 and 16 are available at BeeBase (http://racerx00.tamu.edu/bee_resources.html).
SNPs
SNPs identified from A. m. scutellata whole-genome shotgun (WGS) reads and from the two haplotypes within the A. m. mellifera assembly are available from dbSNP at NCBI (http://www.ncbi.nlm.nih.gov/entrez/ query.fcgi?db=Snp&cmd=Limits) and from the BCM-HGSC ftp site (ftp://ftp.hgsc.bcm.tmc.edu/pub/data/Amellifera/snp). SNPs identified from EST sequences are available from UIUC (http://titan.biotec.uiuc.edu/bee/downloads/bee_downloads.html).
Tiling array data and EST sequences
Tiling array data are available for browsing and download from Systemix (http://www.systemix.org). EST sequences are available from NCBI, DDBJ and EMBL under accessions DB728206–DB781564.
Gene predictions
All of the individual gene sets in the OAIGS, OGS, the community annotated set and BeeBase manually curated set are available for download from the BeeBase downloads page (http://racerx00.tamu.edu/downloadFASTA.html) as either protein or CDS sequences.
References
Wilson, E. O. & Holldobler, B. Eusociality: origin and consequences. Proc. Natl Acad. Sci. USA 102, 13367– 13371 (2005)
Wilson, E. O. The Insect Societies (Harvard Univ. Press, Cambridge, 1971)
Winston, M. L. The Biology of the Honey Bee (Harvard Univ. Press, Cambridge, 1987)
Evans, J. D. & Wheeler, D. E. Gene expression and the evolution of insect polyphenisms. Bioessays 23, 62– 68 (2001)
Page, R. E. & Peng, C. Y. Aging and development in social insects with emphasis on the honey bee, Apis mellifera L. Exp. Gerontol. 36, 695– 711 (2001)
Witthöft, W. Absolute Anzahl und Verteilung der Zellen im Hirn der Honigbiene. Z. Morphol. Tiere 61, 160– 184 (1967)
Menzel, R. Searching for the memory trace in a mini-brain, the honeybee. Learn. Mem. 8, 53– 62 (2001)
von Frisch, K. Dance Language and Orientation of the Honey Bee (Harvard Univ. Press, Cambridge, 1967)
Giurfa, M., Zhang, S., Jenett, A., Menzel, R. & Srinivasan, M. V. The concepts of ‘sameness’ and ‘difference’ in an insect. Nature 410, 930– 933 (2001)
Sheppard, W. S., Rinderer, T. E., Garnery, L. & Shimanuki, H. Analysis of Africanized honey bee mitochondrial DNA reveals further diversity of origin. Genet. Mol. Biol. 22, 73– 75 (1999)
Smith, D. R. & Brown, W. M. Polymorphisms in mitochondrial DNA of European and Africanized honeybees (Apis mellifera). Experientia 44, 257– 260 (1988)
Robinson, G. E., Grozinger, C. M. & Whitfield, C. W. Sociogenomics: social life in molecular terms. Nature Rev. Genet. 6, 257– 270 (2005)
Honey Bee Genome Sequencing Consortium. Proposal for the sequencing of a new target genome: White paper for a honey bee genome project 〈http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/HoneyBee_Genome.pdf〉 (2002)
Morse, R. A. & Calderone, N. W. The value of honey bee pollination in the United States. Bee Culture 128, 1– 15 (2000)
Hamilton, W. D. The genetical evolution of social behaviour, I, II. J. Theor. Biol. 7, 1– 52 (1964)
Hamilton, W. D. Altruism and related phenomena, mainly in social insects. Annu. Rev. Ecol. Syst. 3, 193– 232 (1972)
Crozier, R. H. & Pamilo, P. Evolution of Social Insect Colonies: Sex Allocation and Kin Selection (Oxford Univ. Press, Oxford, 1996)
Foster, K. R., Wenseleers, T. & Ratnieks, F. L. W. Kin selection is the key to altruism. Trends Ecol. Evol. 21, 57– 60 (2006)
Beye, M., Hasselmann, M., Fondrk, M. K., Page, R. E. & Omholt, S. W. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 114, 419– 429 (2003)
Adams, M. D. et al. The genome sequence of Drosophila melanogaster.. Science 287, 2185– 2195 (2000)
Misra, S. et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 3, RESEARCH0083.1– RESEARCH0083.22 (2002)
Richards, S. et al. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 15, 1– 18 (2005)
Holt, R. A. et al. The genome sequence of the malaria mosquito Anopheles gambiae.. Science 298, 129– 149 (2002)
Mongin, E., Louis, C., Holt, R. A., Birney, E. & Collins, F. H. The Anopheles gambiae genome: an update. Trends Parasitol. 20, 49– 52 (2004)
Mita, K. et al. The genome sequence of silkworm, Bombyx mori.. DNA Res. 11, 27– 35 (2004)
Xia, Q. et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306, 1937– 1940 (2004)
Grimaldi, D. & Engel, M. S. Evolution of the Insects (Cambridge Univ. Press, Cambridge, 2005)
Ruttner, F. Biogeography and Taxonomy of Honeybees (Springer, Berlin, 1988)
Garnery, L., Cornuet, J. M. & Solignac, M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1, 145– 154 (1992)
Whitfield, C. W. et al. Thrice out of Africa: ancient and recent expansions of the honey bee, Apis mellifera. Science (in the press).
Havlak, P. et al. The Atlas genome assembly system. Genome Res. 14, 721– 732 (2004)
Solignac, M. et al. A microsatellite-based linkage map of the honeybee, Apis mellifera L. Genetics 167, 253– 262 (2004)
Solignac, M. et al. The genome of Apis mellifera: dialog between linkage mapping and sequence assembly. Genome Biol. (submitted).
Jordan, R. A. & Brosemer, R. W. Characterization of DNA from three bee species. J. Insect Physiol. 20, 2513– 2520 (1974)
Beye, M. & Raeder, U. Rapid DNA preparation from bees and %GC fractionation. Biotechniques 14, 372– 374 (1993)
Human Genome Sequencing Center at Baylor College of Medicine. Honey Bee Genome Project 〈http://www.hgsc.bcm.tmc.edu/projects/honeybee〉 (2006)
Beye, M. et al. Exceptionally high levels of recombination across the honey bee genome. Genome Res. (in the press).
Hunt, G. J. & Page, R. E. Jr. Linkage map of the honey bee, Apis mellifera, based on RAPD markers. Genetics 139, 1371– 1382 (1995)
Beye, M. & Moritz, R. F. Characterization of honeybee (Apis mellifera L.) chromosomes using repetitive DNA probes and fluorescence in situ hybridization. J. Hered. 86, 145– 150 (1995)
Cohen, N., Dagan, T., Stone, L. & Graur, D. GC composition of the human genome: in search of isochores. Mol. Biol. Evol. 22, 1260– 1272 (2005)
Duret, L., Mouchiroud, D. & Gautier, C. Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-rich isochores. J. Mol. Evol. 40, 308– 317 (1995)
Matsuzaki, M. et al. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428, 653– 657 (2004)
Coulondre, C., Miller, J. H., Farabaugh, P. J. & Gilbert, W. Molecular basis of base substitution hotspots in Escherichia coli.. Nature 274, 775– 780 (1978)
Robertson, H. M. & Gordon, K. H. J. Canonical TTAGG repeat telomeres and telomerase in the honey bee, Apis mellifera. Genome Res. (in the press).
Sahara, K., Marec, F. & Traut, W. TTAGG telomeric repeats in chromosomes of some insects and other arthropods. Chromosome Res. 7, 449– 460 (1999)
Fujiwara, H., Osanai, M., Matsumoto, T. & Kojima, K. K. Telomere-specific non-LTR retrotransposons and telomere maintenance in the silkworm, Bombyx mori.. Chromosome Res. 13, 455– 467 (2005)
Biessmann, H. et al. Two distinct domains in Drosophila melanogaster telomeres. Genetics 171, 1767– 1777 (2005)
Casacuberta, E. & Pardue, M. L. HeT-A and TART, two Drosophila retrotransposons with a bona fide role in chromosome structure for more than 60 million years. Cytogenet. Genome Res. 110, 152– 159 (2005)
Melnikova, L. & Georgiev, P. Drosophila telomeres: the non-telomerase alternative. Chromosome Res. 13, 431– 441 (2005)
Biessmann, H., Kobeski, F., Walter, M. F., Kasravi, A. & Roth, C. W. DNA organization and length polymorphism at the 2L telomeric region of Anopheles gambiae.. Insect Mol. Biol. 7, 83– 93 (1998)
Rosen, M. & Edström, J. E. DNA structures common for chironomid telomeres terminating with complex repeats. Insect Mol. Biol. 9, 314– 347 (2000)
Baudry, E. et al. Whole-genome scan in thelytokous-laying workers of the cape honeybee (Apis mellifera capensis): Central fusion, reduced recombination rates and centromere mapping using half-tetrad analysis. Genetics 167, 243– 252 (2004)
Cowan, C. R., Carlton, P. M. & Cande, W. Z. The polar arrangement of telomeres in interphase and meiosis. Rabl organization and the bouquet. Plant Physiol. 125, 532– 538 (2001)
Robertson, H. M. The mariner transposable element is widespread in insects. Nature 362, 241– 245 (1993)
Robertson, H. M. & MacLeod, E. G. Five major subfamilies of mariner transposable elements in insects, including the Mediterranean fruit fly, and related arthropods. Insect Mol. Biol. 2, 125– 139 (1993)
Robertson, H. M. & Lampe, D. J. Distribution of transposable elements in arthropods. Annu. Rev. Entomol. 40, 333– 357 (1995)
Robertson, H. M. in Mobile DNA II (eds Craig, N. L., Craigie, R., Gellert, M. & Lambowitz, A. M.) (ASM, Washington DC, 2002)
Lampe, D. J., Witherspoon, D. J., Soto-Adames, F. N. & Robertson, H. M. Recent horizontal transfer of mellifera subfamily mariner transposons into insect lineages representing four different orders shows that selection acts only during horizontal transfer. Mol. Biol. Evol. 20, 554– 562 (2003)
Ebert, P. R. & Hileman, J. P. T. & Nguyen, H. T. Primary sequence, copy number, and distribution of mariner transposons in the honey bee. Insect Mol. Biol. 4, 69– 78 (1995)
Eiglmeier, K. et al. Comparative analysis of BAC and whole genome shotgun sequences from an Anopheles gambiae region related to Plasmodium encapsulation. Insect Biochem. Mol. Biol. 35, 799– 814 (2005)
Goodwin, T. J., Poulter, R. T., Lorenzen, M. D. & Beeman, R. W. DIRS retroelements in arthropods: identification of the recently active TcDirs1 element in the red flour beetle Tribolium castaneum.. Mol. Genet. Genomics 272, 47– 56 (2004)
Kaminker, J. S. et al. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 3, RESEARCH0084 (2002)
Gillespie, J. J., Johnston, J. S., Cannone, J. J. & Gutell, R. R. Characteristics of the nuclear (18S, 5.8S, 28S, and 5S) and mitochondrial (16S and 12S) rDNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization, and retrotransposable elements. Insect Mol. Biol. (in the press).
Xiong, Y., Burke, W. D., Jakubczak, J. L. & Eickbush, T. H. Ribosomal DNA insertion elements R1Bm and R2Bm can transpose in a sequence specific manner to locations outside the 28S genes. Nucleic Acids Res. 16, 10561– 10573 (1988)
Bigot, Y., Lutcher, F., Hamelin, M. H. & Periquet, G. The 28S ribosomal RNA-encoding gene of Hymenoptera: inserted sequences in the retrotransposon-rich regions. Gene 121, 347– 352 (1992)
Krieger, M. J. & Ross, K. G. Molecular evolutionary analyses of mariners and other transposable elements in fire ants (Hymenoptera: Formicidae). Insect Mol. Biol. 12, 155– 165 (2003)
McAllister, B. F. & Werren, J. H. Phylogenetic analysis of a retrotransposon with implications for strong evolutionary constraints on reverse transcriptase. Mol. Biol. Evol. 14, 69– 80 (1997)
Varricchio, P. et al. Characterization of Aphidius ervi (Hymenoptera, Braconidae) ribosomal genes and identification of site-specific insertion elements belonging to the non-LTR retrotransposon family. Insect Biochem. Mol. Biol. 25, 603– 612 (1995)
Elsik, C. G. et al. Creating a honey bee consensus gene set. Genome Biol. (in the press).
Pearson, W. R. & Lipman, D. J. Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA 85, 2444– 2448 (1988)
Bertone, P. et al. Global identification of human transcribed sequences with genome tiling arrays. Science 306, 2242– 2246 (2004)
Hillier, L. W. et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695– 716 (2004)
Zdobnov, E. M., von Mering, C., Letunic, I. & Bork, P. Consistency of genome-based methods in measuring Metazoan evolution. FEBS Lett. 579, 3355– 3361 (2005)
Bolshakov, V. N. et al. A comparative genomic analysis of two distant diptera, the fruit fly, Drosophila melanogaster, and the malaria mosquito, Anopheles gambiae.. Genome Res. 12, 57– 66 (2002)
Zdobnov, E. M. et al. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster.. Science 298, 149– 159 (2002)
Zdobnov, E. & Bork, P. Quantification of insect genome divergence. Trends Genet. (in the press).
Raible, F. et al. Vertebrate-type intron-rich genes in the marine annelid Platynereis dumerilii.. Science 310, 1325– 1326 (2005)
Ciccarelli, F. D. et al. Complex genomic rearrangements lead to novel primate gene function. Genome Res. 15, 343– 351 (2005)
Tsuchimoto, M. et al. Conservation of novel Mahya genes shows the existence of neural functions common between Hymenoptera and Deuterostome. Dev. Genes Evol. 215, 564– 574 (2005)
Rubin, E. et al. Molecular and phylogenetic analyses reveal mammalian-like clockwork in the honey bee (Apis mellifera) and shed new light on the molecular evolution of the circadian clock. Genome Res. 16(11), 1352– 1365 (2006)
Velarde, R. A., Sauer, C. D., Walden, K. K., Fahrbach, S. E. & Robertson, H. M. Pteropsin: A vertebrate-like non-visual opsin expressed in the honey bee brain. Insect Biochem. Mol. Biol. 35, 1367– 1377 (2005)
Pires-daSilva, A. & Sommer, R. J. The evolution of signalling pathways in animal development. Nature Rev. Genet. 4, 39– 49 (2003)
Dearden, P. K. et al. Patterns of conservation and change in honeybee developmental genes. Genome Res. (in the press).
Tribolium Genome Sequencing Project. Tribolium castaneum v 2. 0 assembly 〈http://www.hgsc.bcm.tmc.edu/projects/tribolium〉 (2006)
Lehmann, R. & Nusslein-Volhard, C. Abdominal segmentation, pole cell formation, and embryonic polarity require the localized activity of oskar, a maternal gene in Drosophila.. Cell 47, 141– 152 (1986)
Schroder, R. vasa mRNA accumulates at the posterior pole during blastoderm formation in the flour beetle Tribolium castaneum.. Dev. Genes Evol. 216, 277– 283 (2006)
Nakao, H. Isolation and characterization of a Bombyx vasa-like gene. Dev. Genes Evol. 209, 312– 316 (1999)
Dearden, P. K. Germ cell development in the Honeybee (Apis mellifera); Vasa and Nanos expression. BMC Dev. Biol. 6 doi: 10.1186/1471-213X-6-6 (17 February (2006)
Wilkins, A. S. The Evolution of Developmental Pathways (Sinauer, Sunderland, Massachusetts, 2002)
Davidson, E. H. Genomic Regulatory Systems (Academic, London, 2001)
Cline, T. W. & Meyer, B. J. Vive la difference: males vs females in flies vs worms. Annu. Rev. Genet. 30, 637– 702 (1996)
Albert, S., Bhattacharya, D., Klaudiny, J., Schmitzova, J. & Simuth, J. The family of major royal jelly proteins and its evolution. J. Mol. Evol. 49, 290– 297 (1999)
Drapeau, M. D., Albert, S., Kucharski, R., Prusko, C. & Maleszka, R. Evolution of the Yellow/Major Royal Jelly Protein family and the emergence of sex-specific social behavior in honeybees. Genome Res. (in the press).
Evans, J. D. & Wheeler, D. E. Differential gene expression between developing queens and workers in the honey bee, Apis mellifera.. Proc. Natl Acad. Sci. USA 96, 5575– 5580 (1999)
Evans, J. D. & Wheeler, D. E. Expression profiles during honeybee caste determination. Genome Biol. 2, RESEARCH0001 (2000)
Corona, M., Estrada, E. & Zurita, M. Differential expression of mitochondrial genes between queens and workers during caste determination in the honeybee Apis mellifera.. J. Exp. Biol. 202, 929– 938 (1999)
Hepperle, C. & Hartfelder, K. Differentially expressed regulatory genes in honey bee caste development. Naturwissenschaften 88, 113– 116 (2001)
West-Eberhard, M. J. Developmental Plasticity and Evolution (Oxford Univ. Press, New York, 2003)
Colombani, J. et al. Antagonistic actions of ecdysone and insulins determine final size in Drosophila.. Science 310, 667– 670 (2005)
Mirth, C., Truman, J. W. & Riddiford, L. M. The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster.. Curr. Biol. 15, 1796– 1807 (2005)
Willis, J. H., Ionomidou, V. A., Smith, R. F. & Hamodrakas, S. J. in Comprehensive Molecular Insect Science (eds Gilbert, L. I., Iatrou, K. & Gill, S. S.) 79– 110 (Elsevier, Oxford, 2005)
Tellam, R. L., Wijffels, G. & Willadsen, P. Peritrophic matrix proteins. Insect Biochem. Mol. Biol. 29, 87– 101 (1999)
Jones, A. K., Raymond-Delpech, V., Thany, S. H., Gauthier, M. & Sattelle, D. B. The nicotinic acetylcholine receptor gene family of the honeybee, Apis mellifera. Genome Res. (in the press).
Eisenhardt, D., Kühn, C. & Leboulle, G. The PKA-CREB system encoded by the honeybee genome. Insect Mol. Biol. (in the press).
Hummon, A. B. et al. From the genome to the proteome: Uncovering peptides in the Apis brain. Science (in the press).
Baggerman, G., Cerstiaens, A., De Loof, A. & Schoofs, L. Peptidomics of the larval Drosophila melanogaster central nervous system. J. Biol. Chem. 277, 40368– 40374 (2002)
Hewes, R. S. & Taghert, P. H. Neuropeptides and neuropeptide receptors in the Drosophila melanogaster genome. Genome Res. 11, 1126– 1142 (2001)
Riehle, M. A., Garczynski, S. F., Crim, J. W., Hill, C. A. & Brown, M. R. Neuropeptides and peptide hormones in Anopheles gambiae.. Science 298, 172– 175 (2002)
Hauser, F., Cazzamali, G., Williamson, M., Blenau, W. & Grimmelikhuijzen, C. J. P. A review of neurohormone GPCRs present in the fruitfly Drosophila melanogaster and the honey bee Apis mellifera.. Prog. Neurobiol. 80, 1– 19 (2006)
Clyne, P. J. et al. A novel family of divergent seven-transmembrane proteins: candidate odorant receptors in Drosophila.. Neuron 22, 327– 338 (1999)
Robertson, H. M., Warr, C. G. & Carlson, J. R. Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster.. Proc. Natl Acad. Sci. USA 100, (suppl. 2) 14537– 14542 (2003)
Vosshall, L. B., Amrein, H., Morozov, P. S., Rzhetsky, A. & Axel, R. A spatial map of olfactory receptor expression in the Drosophila antenna. Cell 96, 725– 736 (1999)
Hill, C. A. et al. G-protein-coupled receptors in Anopheles gambiae.. Science 298, 176– 178 (2002)
Robertson, H. M. & Wanner, K. W. The chemoreceptor superfamily in the honey bee Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res. (in the press).
Galizia, C. G. & Menzel, R. The role of glomeruli in the neural representation of odours: results from optical recording studies. J. Insect Physiol. 47, 115– 130 (2001)
Fishilevich, E. & Vosshall, L. B. Genetic and functional subdivision of the Drosophila antennal lobe. Curr. Biol. 15, 1548– 1553 (2005)
Clyne, P. J., Warr, C. G. & Carlson, J. R. Candidate taste receptors in Drosophila.. Science 287, 1830– 1834 (2000)
Xu, P., Atkinson, R., Jones, D. N. & Smith, D. P. Drosophila OBP LUSH is required for activity of pheromone-sensitive neurons. Neuron 45, 193– 200 (2005)
Foret, S. & Maleszka, R. Function and evolution of odorant binding protein gene family in a social insect, the honey bee (Apis mellifera). Genome Res. (in the press).
Panda, S., Hogenesch, J. B. & Kay, S. A. Circadian rhythms from flies to human. Nature 417, 329– 335 (2002)
Guillaumond, F., Dardente, H., Giguere, V. & Cermakian, N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J. Biol. Rhythms 20, 391– 403 (2005)
Morse, R. A. & Flottum, K. (eds). Honey Bee Pests, Predators and Diseases (A. I. Root Co., Medina, Ohio, 1997)
Schmid-Hempel, P. Parasites in Social Insects (Princeton Univ. Press, Princeton, New Jersey, 1998)
Evans, J. D. et al. Immune pathways and defence mechanisms in honey bees. Insect Mol. Biol. (in the press).
Aronstein, K. & Saldivar, E. Characterization of a honey bee Toll related receptor gene Am18w and its potential involvement in antimicrobial immune defense. Apidologie (Celle) 36, 3– 14 (2005)
Evans, J. D. Beepath: An ordered quantitative-PCR array for exploring honey bee immunity and disease. J. Invertebr. Pathol. 93, 135– 139 (2006)
Evans, J. D. & Pettis, J. S. Colony-level effects of immune responsiveness in honey bees, Apis mellifera.. Evol. Int. J. Org. Evol. 59, 2270– 2274 (2005)
Chen, Y. P., Higgins, J. A. & Feldlaufer, M. F. Quantitative real-time reverse transcription-PCR analysis of deformed wing virus infection in the honeybee (Apis mellifera L.). Appl. Environ. Microbiol. 71, 436– 441 (2005)
Gregorc, A. & Bowen, I. D. Histopathological and histochemical changes in honeybee larvae (Apis mellifera L.) after infection with Bacillus larvae, the causative agent of American foulbrood disease. Cell Biol. Int. 22, 137– 144 (1998)
Bogdanov, S., Kilchenmann, V. & Imdorf, A. Acaricide residues in some bee products. J. Apicultural. Res. 37, 57– 67 (1998)
Tremolada, P., Bernardinelli, I., Colombo, M., Spreafico, M. & Vighi, M. Coumaphos distribution in the hive ecosystem: case study for modeling applications. Ecotoxicology 13, 589– 601 (2004)
Wallner, K. The use of varroacides and their influence on the quality of bee products. Am. Bee J. 135, 817– 821 (1995)
Berenbaum, M. R. in Molecular Biology of the Toxic Response (eds Puga, A. & Wallace, K. B.) 553– 571 (Taylor & Francis, Philadelphia, 1999)
Oakeshott, J. G., Claudianos, C., Campbell, P. M., Newcomb, R. D. & Russell, R. J. in Comprehensive Molecular Insect Science (eds Gilbert, L. I., Iatrou, K. & Gill, S.) 309– 381 (Elsevier Pergamon, Oxford, 2005)
Ranson, H. & Hemingway, J. in Comprehensive Molecular Insect Science (eds Gilbert, L. I., Iatrou, K. & Gill, S.) 383– 402 (Elsevier Pergamon, Oxford, 2005)
Feyereisen, R. in Comprehensive Molecular Insect Science (eds Gilbert, L. I., Iatrou, K. & Gill, S.) 1– 77 (Elsevier Pergamon, Oxford, 2005)
Ranson, H. et al. Evolution of supergene families associated with insecticide resistance. Science 298, 179– 181 (2002)
Claudianos, C. et al. A deficit of detoxification enzymes: Pesticide sensitivity and environmental response in the honeybee. Insect Mol. Biol. (in the press).
Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6– 21 (2002)
Wang, Y. et al. Functional CpG methylation system in a social insect. Science (in the press).
Winston, W. M., Molodowitch, C. & Hunter, C. P. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science 295, 2456– 2459 (2002)
Beye, M., Hartel, S., Hagen, A., Hasselmann, M. & Omholt, S. W. Specific developmental gene silencing in the honey bee using a homeobox motif. Insect Mol. Biol. 11, 527– 532 (2002)
Amdam, G. V., Simoes, Z. L., Guidugli, K. R., Norberg, K. & Omholt, S. W. Disruption of vitellogenin gene function in adult honeybees by intra-abdominal injection of double-stranded RNA. BMC Biotechnol. 3 doi: 10.1186/1472-6750-3-1 (20 January (2003)
Aronstein, K., Pankiw, T. & Saldivar, E. SID-1 is implicated in systemic gene silencing in the honey bee. J. Apic. Res. 45, 20– 24 (2006)
Thummel, C. S. From embryogenesis to metamorphosis: the regulation and function of Drosophila nuclear receptor superfamily members. Cell 83, 871– 877 (1995)
Velarde, R. A., Fahrbach, S. & Robinson, G. E. Nuclear receptors of the honey bee: annotation and expression in the adult brain. Insect Mol. Biol. (in the press).
Gerber, S. et al. The photoreceptor cell-specific nuclear receptor gene (PNR) accounts for retinitis pigmentosa in the Crypto-Jews from Portugal (Marranos), survivors from the Spanish Inquisition. Hum. Genet. 107, 276– 284 (2000)
Kobayashi, M. et al. Identification of a photoreceptor cell-specific nuclear receptor. Proc. Natl Acad. Sci. USA 96, 4814– 4819 (1999)
Sinha, S., Ling, X., Whitfield, C. W., Zhai, C. & Robinson, G. E. Genome scan for cis-regulatory DNA motifs associated with social behavior in honey bees. Proc. Natl Acad. Sci. USA (in the press).
Whitfield, C. W. et al. Genomic dissection of behavioural maturation in the honey bee. Proc. Natl Acad. Sci. USA (in the press).
Whitfield, C. W., Cziko, A. M. & Robinson, G. E. Gene expression profiles in the brain predict behavior in individual honey bees. Science 302, 296– 299 (2003)
Savard, J. et al. Phylogenomic analysis reveals bees and wasps (Hymenoptera) at the base of the radiation of holometabolous insects. Genome Res. (in the press).
Danforth, B. N., Fang, J. & Sipes, S. Analysis of family-level relationships in bees (Hymenoptera: Apiformes) using 28S and two previously unexplored nuclear genes: CAD and RNA polymerase II. Mol. Phylogen. Evol. 39, 358– 372 (2006)
Danforth, B. N., Sipes, S., Fang, J. & Brady, S. G. T he history of early bee diversification based on five genes plus morphology. Proc. Natl Acad. Sci. USA. doi:10.1073/pnas.0604033103 (2 October 2006)
Marth, G. T. et al. A general approach to single-nucleotide polymorphism discovery. Nature Genet. 23, 452– 456 (1999)
Ruttner, F., Tassencourt, L. & Louveaux, J. Biometric-statistical analysis of the geographical variability of Apis mellifera L. I. Material and methods. Apidologie (Celle) 9, 363– 381 (1978)
Franck, P., Garnery, L., Solignac, M. & Cornuet, J. M. Molecular confirmation of a fourth lineage in honeybees from the Near East. Apidologie (Celle) 31, 167– 180 (2000)
Sheppard, W. S., Rinderer, T. E., Mazzoli, J. A., Stelzer, J. A. & Shimanuki, H. Gene flow between African- and European-derived honey bee populations in Argentina. Nature 349, 782– 784 (1991)
Iltis, H. Life of Mendel (W. W. Norton and Company, Inc., New York, 1923)
Wang, J. et al. SilkDB: a knowledgebase for silkworm biology and genomics. Nucleic Acids Res. 33, D399– D402 (2005)
Robertson, H. M. Insect genomes. Am. Entomol. 51, 166– 171 (2005)
Hunt, G. et al. Behavioral genomics of honeybee foraging and nest defence. Naturwissenschaften (in the press).
Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69– 87 (2005)
Sarfare, S., Ahmad, S. T., Joyce, M. V., Boggess, B. & O’Tousa, J. E. The Drosophila ninaG oxidoreductase acts in visual pigment chromophore production. J. Biol. Chem. 280, 11895– 11901 (2005)
Ahmad, S. T., Joyce, M. V., Boggess, B. & O’Tousa, J. E. The role of Drosophila ninaG oxidoreductase in visual pigment chromophore biogenesis. J. Biol. Chem. 281, 9205– 9209 (2006)
Smith, W. C. & Goldsmith, T. H. Phyletic aspects of the distribution of 3-hydroxyretinal in the class insecta. J. Mol. Evol. 30, 72– 84 (1990)
Song, J., Wu, L., Chen, Z., Kohanski, R. A. & Pick, L. Axons guided by insulin receptor in Drosophila visual system. Science 300, 502– 505 (2003)
Shieh, B. H., Zhu, M. Y., Lee, J. K., Kelly, I. M. & Bahiraei, F. Association of INAD with NORPA is essential for controlled activation and deactivation of Drosophila phototransduction in vivo.. Proc. Natl Acad. Sci. USA 94, 12682– 12687 (1997)
Townson, S. M. et al. Honeybee blue- and ultraviolet-sensitive opsins: cloning, heterologous expression in Drosophila, and physiological characterization. J. Neurosci. 18, 2412– 2422 (1998)
Skrzipek, K-H. & Skrzipek, H. The ninth retinula cell in the ommatidium of the worker honey bee (Apis mellifica L.). Z. Zellforsch. Mikrosk. Anat. 147, 589– 593 (1974)
Tomlinson, A. & Struhl, G. Delta/Notch and Boss/Sevenless signals act combinatorially to specify the Drosophila R7 photoreceptor. Mol. Cell 7, 487– 495 (2001)
Lee, Y. et al. Pyrexia is a new thermal transient receptor potential channel endowing tolerance to high temperatures in Drosophila melanogaster.. Nature Genet. 37, 305– 310 (2005)
Rosenzweig, M. et al. The Drosophila ortholog of vertebrate TRPA1 regulates thermotaxis. Genes Dev. 19, 419– 424 (2005)
Tracey, W. D., Wilson, R. I., Laurent, G. & Benzer, S. painless, a Drosophila gene essential for nociception. Cell 113, 261– 273 (2003)
O’Hagan, R., Chalfie, M. & Goodman, M. B. The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nature Neurosci. 8, 43– 50 (2005)
Si, A., Helliwell, P. & Maleszka, R. Effects of NMDA receptor antagonists on olfactory learning and memory in the honeybee (Apis mellifera). Pharmacol. Biochem. Behav. 77, 191– 197 (2004)
Xia, S. et al. NMDA receptors mediate olfactory learning and memory in Drosophila.. Curr. Biol. 15, 603– 615 (2005)
Kucharski, R., Ball, E. E., Hayward, D. C. & Maleszka, R. Molecular cloning and expression analysis of a cDNA encoding a glutamate transporter in the honeybee brain. Gene 242, 399– 405 (2000)
Zhu, H., Yuan, Q., Froy, O., Casselman, A. & Reppert, S. M. The two CRYs of the butterfly. Curr. Biol. 15, R953– R954 (2005)
Gaunt, M. W. & Miles, M. A. An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol. Biol. Evol. 19, 748– 761 (2002)
Nei, M. Genetic distance between populations. Am. Nat. 106, 283– 292 (1972)
Acknowledgements
Work at the BCM-HGSC was supported by grants from the NHGRI and USDA. BAC and fosmid Library construction was supported by a subcontract from a grant awarded to J.S.J., organized by D.B.W. (President, Bee Weaver Apiaries, Inc.). Fgenesh and Fgenesh++ analysis was donated by Softberry. Other support was received from NIH NIAID (H.M.R.), NRI Functional Genomics (G.E.R.) and Illinois Sociogenomics Initiative (G.E.R.), NSF (M.M.E.; B. Schatz, UIUC), Intramural Research Program NIH NLM (R.A.), USDA-NRI, California Beekeepers Assoc., Texas Beekeepers Assoc., T. W. Burleson and Son, Inc., TAMU, NIH NLM (J.G.R.), RSNZ Marsden Fund (P.K.D.), Danish Research Agency, Carlsberg Foundation, Novo Nordisk Foundation, and DFG. The authors thank the production staff at the HGSC.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Correspondence and requests for materials should be addressed to G.M.W. gwstock@bcm.edu.
Author information
Authors and Affiliations
Consortia
Supplementary information
Supplementary Notes
This file contains Supplementary Figure, Supplementary Tables and Supplementary Methods. (PDF 3694 kb)
Rights and permissions
About this article
Cite this article
The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949 (2006). https://doi.org/10.1038/nature05260
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature05260
This article is cited by
-
Mechanisms for polyandry evolution in a complex social bee
Behavioral Ecology and Sociobiology (2024)
-
Foraging Activity of Honey Bees (Apis mellifera L., 1758) and Exposure to Cadmium: a Review
Biological Trace Element Research (2024)
-
Intrasexual cuticular hydrocarbon dimorphism in a wasp sheds light on hydrocarbon biosynthesis genes in Hymenoptera
Communications Biology (2023)
-
Putting hornets on the genomic map
Scientific Reports (2023)
-
In silico SNP prediction of selected protein orthologues in insect models for Alzheimer's, Parkinson's, and Huntington’s diseases
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
