
Abstract
The influenza pandemic of 1918–19 was responsible for about 50 million deaths worldwide1. Modern histopathological analysis of autopsy samples from human influenza cases from 1918 revealed significant damage to the lungs with acute, focal bronchitis and alveolitis associated with massive pulmonary oedema, haemorrhage and rapid destruction of the respiratory epithelium2. The contribution of the host immune response leading to this severe pathology remains largely unknown. Here we show, in a comprehensive analysis of the global host response induced by the 1918 influenza virus, that mice infected with the reconstructed 1918 influenza virus displayed an increased and accelerated activation of host immune response genes associated with severe pulmonary pathology. We found that mice infected with a virus containing all eight genes from the pandemic virus showed marked activation of pro-inflammatory and cell-death pathways by 24 h after infection that remained unabated until death on day 5. This was in contrast with smaller host immune responses as measured at the genomic level, accompanied by less severe disease pathology and delays in death in mice infected with influenza viruses containing only subsets of 1918 genes. The results indicate a cooperative interaction between the 1918 influenza genes and show that study of the virulence of the 1918 influenza virus requires the use of the fully reconstructed virus. With recent concerns about the introduction of highly pathogenic avian influenza viruses into humans and their potential to cause a worldwide pandemic with disastrous health and economic consequences, a comprehensive understanding of the global host response to the 1918 virus is crucial. Moreover, understanding the contribution of host immune responses to virulent influenza virus infections is an important starting point for the identification of prognostic indicators and the development of novel antiviral therapies.
Similar content being viewed by others

Main
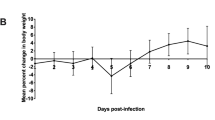
To study the virulence and pathogenesis of the 1918 influenza virus, mice were infected intranasally with a contemporary human influenza A/Texas/36/91 H1N1 virus (Tx91), Tx91 virus containing the 1918 HA and NA genes (2:6 1918), the 1918 HA, NA, M, NP and NS genes (5:3 1918), or the reconstructed 1918 (H1N1) recombinant virus (r1918). Mice infected with r1918 showed the most severe weight loss and the earliest mean time of death of all viruses tested (Supplementary Information). Moreover, titration of virus recovered from lung tissue showed that mice infected with r1918 had about tenfold higher levels of virus on days 1 and 3 after infection than those infected with 2:6 1918 or 5:3 1918. By day 5 after infection there was a slight decrease in the lung titres of r1918-infected mice, and mice infected with 2:6 1918, 5:3 1918 and r1918 showed similar lung titres of about 107 50% embryo-infective doses per ml. Our results were consistent with those in previous reports3: most mice infected with r1918 were dead by day 5 after infection.
Previous studies showed increased lung pathology in mice infected with influenza viruses bearing genes from the 1918 virus3,4,5. A more detailed histological analysis of mice infected with 5:3 1918, 2:6 1918 or r1918 viruses revealed differences in pathology during the course of infection. Whereas mice infected with Tx91 reassortant virus had minimal to mild diffuse histiocytic alveolitis without neutrophils or lesions in the bronchi or bronchioles (Fig. 1a), mice infected with 5:3 1918, 2:6 1918 or r1918 viruses had pulmonary lesions that consisted of mild to severe necrotizing bronchitis with accompanying moderate to severe alveolitis and associated moderate to severe alveolar oedema (Fig. 1b–f). However, the character of the alveolitis varied between the 1918 viruses and the sampling periods. At day 3, the r1918-infected mice had acute moderate to severe diffuse alveolitis with a prominent neutrophilic component and severe alveolar oedema (Fig. 1b). For some lungs, alveolitis was multifocal with intense neutrophilic infiltration that distended the alveoli (Fig. 1c). By contrast, mice infected with 5:3 1918 and 2:6 1918 viruses had moderate to severe alveolitis, but histiocytes predominated over neutrophils and the alveolar oedema was only moderately severe (Fig. 1d, e). At day 5 the pulmonary response in r1918-infected mice had changed to subacute and was more severe with diffuse alveolitis retaining the severe oedema, but changing to a predominance of histiocytes with fewer neutrophils (Fig. 1f). The bronchial and bronchiolar lesions were similar to those at day 3. However, in some r1918-infected lungs, an acute inflammatory response continued, as was evident from multiple scattered foci of intense neutrophilic alveolitis. In contrast, some mice infected with 5:3 1918 and 2:6 1918 viruses showed slightly less alveolitis, alveolar oedema, bronchitis and peribronchitis on day 5 than on day 3. These results show that the pathology induced by r1918 was distinct from that observed in mice infected with 5:3 1918 and 2:6 1918 viruses, indicating that cooperation between the individual 1918 viral proteins might be required for the induction of maximum pathology.

a, Mild histiocytic alveolitis with normal bronchiole, Tx/91 virus, day 3. b, Moderate diffuse alveolitis, predominantly neutrophilic, but with macrophages and associated haemorrhage and oedema, r1918 virus, day 3. c, Severe focal alveolitis with neutrophils distending affected alveoli, r1918 virus, day 3. d, e, Moderate histiocytic alveolitis and some neutrophils and oedema, 5:3 1918 (d) and 2:6 1918 (e) viruses, day 3. f, Severe diffuse histiocytic alveolitis with severe oedema, r1918 virus, day 5. Scale bar, 20 µm.
To understand why 1918 influenza virus showed extreme virulence in mice, we used gene expression analysis to provide a global view of the host response in lungs of infected mice. Expression microarray analysis was performed by taking equal masses of total RNA isolated from lungs of mice infected with strain Tx91, 2:5 1918, 5:3 1918 or r1918 and comparing them with a common reference sample prepared from lungs of mock-infected mice. Lungs from three mice were harvested for each virus on days 1, 3 and 5 after infection (total n = 36). Initially, our analysis was directed to genes associated with the activation of key immune cells known to be important for the response to influenza virus infection, including T helper 1 (TH1), natural killer, macrophage and neutrophil cells (shown in Fig. 2). This analysis revealed a significant increase in the expression of genes associated with these immune cell populations in lungs of mice infected with r1918. Expression patterns of cytokines, chemokines and apoptosis-related genes showed the most significant and earliest activation during r1918 infection (Supplementary Information). The increased expression of immune-cell-related genes was correlated with virulence, because strain r1918 induced the most significant and earliest expression of immune-response-related genes, followed in magnitude and occurrence by 2:6 1918, 5:3 1918 and Tx91. Because many innate response-related genes showed saturation of the fluorescence signal by expression microarray, we measured the messenger RNA and protein expression of several key cytokines, including tumour necrosis factor (TNF), by quantitative real-time polymerase chain reaction (PCR) and enzyme-linked immunosorbent assay (ELISA). This analysis demonstrated that r1918 infections consistently resulted in a higher and prolonged expression of these mRNAs and proteins (Supplementary Information). It was shown recently6 that increased production of cytokines and chemokines during infection with 2:6 1918 could be attributed to the presence of neutrophils and alveolar macrophages in the lungs of infected mice. Our studies showed that r1918 infections also resulted in increased neutrophil and macrophage recruitment, accompanied by a significant increase in the expression of cytokine and chemokine genes. Expression of genes related to T helper 2 (TH2) cell and nitric oxide metabolism showed similar patterns in all viral infections on days 1, 3 and 5, indicating that these responses might not have been primary mediators of r1918 virulence (Supplementary Information). The cellular origins of the genes identified by microarray are unknown, but taken together these data show that r1918 produced the most severe and rapid disease in mice, which could be correlated with an increased expression of inflammatory response genes similar to that reported for highly pathogenic avian influenza viruses7,8.

Expression of selected immune-cell-related genes in the lungs of mice infected with Tx91, 2:6 1918, 5:3 1918 or r1918 viruses on days 1, 3 and 5 after infection, as determined by expression microarray analysis. For each infection point, the data presented are the error-weighted average expression changes calculated from four technical replicate arrays performed on three individual mice (n = 12 total). Genes shown in red were upregulated and genes shown in green were downregulated in infected relative to mock-infected mouse lung. NK, natural killer; TH1, T helper 1.
To characterize the functional consequences of gene expression changes associated with infection with r1918, we performed pathway analysis of the gene expression data with Ingenuity Pathways Analysis (see Methods). As shown in Fig. 3, pathway analysis of the expression array data revealed that infection with r1918 virus resulted in the most significant activation of death receptor, interleukin-6, type I interferon and Toll-like receptor responses. These data show that the aggregate host response to r1918 virus infection results in an increase in pro-inflammatory and cell death responses. We observed an inverse correlation between influenza virulence and G2/M checkpoint control and glutathione metabolism. The increased expression of cell-cycle and glutathione metabolism genes in Tx91 infections could be important components in the successful clearance of influenza virus infection and tissue remodelling without severe immunopathology. We also suggest that activation of these responses reflects the importance of protecting cells from damage by oxygen radicals and the need for the proliferation and repopulation of damaged or killed cells in the respiratory epithelium.

a–d, Analysis of the biological response pathways that showed most significant activation during r1918 infection, including death receptor (a), interleukin-6 (b), interferon (c) and Toll-like receptor (d) pathways. e, f, The biological responses most significantly induced in the non-lethal Tx91 infections: G2/M checkpoint control (e) and glutathione metabolism (f). Pathway analysis was performed on genes that were induced more than twofold (P < 0.01) in any of the infections compared with uninfected controls. For this purpose, Ingenuity Pathway Analysis was used, which analyses the RNA expression data in the context of known biological response and regulatory networks, as well as other higher-order response pathways to assign functional information and biological relevance. These results are shown as the negative logarithm of significance, which is a statistical score and is a measure of the likelihood of the genes in a given network being found together as a result of chance, as determined by Fisher's exact test (see Methods for details).
To explore the host responses associated with the severe immunopathology of infections with r1918 virus, we characterized the functional relationship between genes that were induced more than twofold (P < 0.01) only in r1918 virus infections. As shown in Fig. 4, many of these genes that were uniquely induced more than twofold in r1918-infected mouse lung compared with controls were involved in cell death and inflammatory responses and have been reported to have direct and/or indirect interactions. Infections with r1918 resulted in a statistically significant coordinated activation of multiple cell-death-related genes, particularly those related to death receptor responses. This is significant because activation of TNF-related pathways has been shown to be associated with H5N1 influenza viral infections of macrophages9. Moreover, r1918 infection-induced expression of mRNAs for Fas, caspase-8 and caspase-9 is significant because of the central roles of these proteins in the induction of apoptosis through the death-receptor and mitochondrial apoptosis pathways. Caspase-9 is activated by the release of cytochrome c from permeabilized mitochondria, and it has been reported that an influenza viral protein expressed from an alternative reading frame of the PB1 gene segment (PB1-F2) is trafficked to mitochondria and can induce apoptosis10. It has recently been reported11 that PB1-F2 associates with components of the permeability transition pore complex and sensitizes cells to apoptotic stimuli, including TNF-α, resulting in mitochondrial permeabilization and the release of cytochrome c. We believe our data showing increased cell death responses in r1918-infected mice may be related to the effects of the 1918 PB1-F2 protein. However, these observations do not rule out additional functions of the 1918 polymerase proteins, because these genes are also important virulence factors for highly pathogenic avian influenza viruses12,13,14,15,16,17.

Biological network of selected genes that were only induced more than twofold (P < 0.01) in the r1918-infected mouse lung compared with uninfected controls. This diagram shows the direct (solid lines) and indirect (dashed lines) interactions reported for these cell-death-related (blue shading) and immune-response-related (yellow shading) genes; grey denotes genes with multiple and/or undefined biological functions. Biological network analysis was performed by using Ingenuity Pathway Analysis and showed statistical significance with Fisher's exact test to calculate a P-value determining the probability that each biological function and/or disease assigned to that data set was due to chance alone (see Methods for details).
Taken together, our results indicate that enhanced inflammatory and cell death responses might be contributors to severe immunopathology associated with r1918 influenza virus infections, although it is not possible to completely exclude the possibility that increased immune and cell death responses may be a consequence rather than the cause of enhanced pathology of r1918 infection. Our study also shows the synergistic effect of the 1918 gene segments and that exceptional virulence and exaggerated host global gene expression response may be dependent on the complete constellation of 1918 genes. We acknowledge that a more complete understanding of r1918 virulence will require experiments in additional animal models, including ferrets, pigs and non-human primates. Characterization of the global host response to other highly virulent viruses will be important in understanding the role of the host in influenza virus pathology, and we are currently comparing these results with findings from similar studies of animals infected with highly pathogenic avian influenza viruses.
Methods
Recombinant influenza viruses
Recombinant viruses were generated with a reverse genetics system18. The low cell (C1 or C2) passage influenza A H1N1 virus stocks were grown in Madin–Darby canine kidney cells. The reconstructed strain of human influenza H1N1 A/Texas/36/91 (Tx/91) virus has been characterized previously3 and causes common influenza symptoms in humans in experimental settings19. The r1918 influenza virus genes correspond to the published sequence of influenza A/South Carolina/1/18 (H1N1) virus HA open reading frame20, and influenza A/Brevig Mission/1/18 (H1N1) virus NA, M, NS, NP, PA, PB1 and PB2 open reading frames12,21,22,23,24. The 1918 recombinant viruses contain the 5′ and 3′ non-coding regions corresponding to influenza A/WSN/33 (H1N1) virus.
Mouse experiments
Female BALB/c mice (Harlan Laboratories) 6–7 weeks old were anesthetized with ketamine–xylazine (1.98 and 0.198 mg per mouse, respectively) and inoculated intranasally with 106 plaque-forming units of the indicated virus. Because of the virulence of r1918 in mice, increased numbers of animals were infected with r1918 to allow the collection of samples at day 5 after infection3. Lungs were harvested at days 1, 3 and 5 after infection for viral titration and histopathology, and extraction of total RNA was performed from a 10% (w/v) lung homogenate as described previously5. All animal work was performed in a specially separated negative-pressure HEPA (high-efficiency particulate air)-filtered biosafety level laboratory (BSL3) with enhancements. All personnel wore half-body Racal hoods with backpack HEPA-filtered air supplies3.
Histopathology
Three mice from each group were killed at days 3 and 5 after infection A 5-mm lung piece from the ventral end of each left lobe was collected for histopathology. Tissues were fixed with standard procedures in 10% neutral-buffered formalin solution, sectioned, and stained with haematoxylin and eosin.
Expression microarray analysis and bioinformatics
Total RNA isolation and mRNA amplification were performed on equal masses of total RNA isolated from lungs of infected mice as described previously25 and were compared with an equal-mass pool of RNA from lungs of ten individual mock-infected mice. For each infection group, expression oligonucleotide arrays (n = 4 per gene) were performed on RNA isolated from lung tissue from three individual animals (n = 12 total). The data presented are the error-weighted average changes in expression calculated from four technical replicate arrays performed on three individual mice (n = 12 final for each gene). Probe labelling and microarray slide hybridization were performed with mouse oligonucleotide arrays (G4121A; Agilent Technologies)25. All data were entered into a custom-designed Oracle 9i backed relational database, Expression Array Manager, and were then uploaded into Rosetta Resolver System 5.0 (Rosetta Biosoftware) and Spotfire Decision Site 8.1 (Spotfire). All primary expression microarray data are available at the University of Washington's Public Microarray Data Download site (http://expression.microslu.washington.edu), in accordance with the proposed MIAME (minimum information about a microarray experiment) standards26.
Functional analysis of statistically significant gene expression changes was performed with Ingenuity Pathways Analysis (Ingenuity Systems). This software analyses RNA expression data in the context of known biological response and regulatory networks as well as other higher-order response pathways. Ingenuity functional analysis identified biological functions and/or diseases that were most significant. Genes from the data set that met the twofold (P < 0.01) change cutoff and were associated with biological functions in the Ingenuity Pathways Knowledge Base were considered for analysis. For all analyses, Fisher's exact test was used to calculate a P-value determining the probability that each biological function assigned to that data set was due to chance alone. In the functional network shown in Fig. 4, genes are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). All edges are supported by at least one published reference or from canonical information stored in the Ingenuity Pathways Knowledge Base. For these analyses, Fisher's exact test was used to calculate a P-value determining the probability that each biological function and/or disease assigned to that data set was due to chance alone.
Real-time PCR assays
Quantitative real-time PCR was used to validate changes in gene expression25. Primer and probe sets for each of the target sequences were chosen from the Applied Biosystems Assays-on-Demand product list. Quantification of each gene, relative to the calibrator, was performed with the equation  within the Applied Biosystems Sequence Detections Software version 1.2.2.
within the Applied Biosystems Sequence Detections Software version 1.2.2.
Cytokine and chemokine quantification
To determine levels of cytokine or chemokine proteins in lung, at each time point examined three mice from each group were exsanguinated from the axilla and killed; lung tissues were then removed from naive and infected mice (days 1, 3 and 5 after infection). Individual whole lung samples were immediately frozen at -70 °C. At the time of analysis, tissues were thawed, homogenized in 1 ml of cold PBS and centrifuged at 150g for 5 min. With the use of ELISA kits from R&D Systems, the clarified cell lysates were assayed for mouse IP-10 (CXCL10), TNF-α, macrophage inflammatory protein (MIP)-1α (CCL3) and MIP-2 (CXCL2).
References
Johnson, N. P. & Mueller, J. Updating the accounts: global mortality of the 1918–1920 ‘Spanish’ influenza pandemic. Bull. Hist. Med. 76, 105–115 (2002)
Taubenberger, J. K. & Morens, D. M. 1918 influenza: the mother of all pandemics. Emerg. Infect. Dis. 12, 15–22 (2006)
Tumpey, T. M. et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310, 77–80 (2005)
Kobasa, D. et al. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431, 703–707 (2004)
Kash, J. C. et al. Global host immune response: pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. J. Virol. 78, 9499–9511 (2004)
Tumpey, T. M. et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79, 14933–14944 (2005)
Zhou, J. et al. Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses. J. Infect. Dis. 194, 61–70 (2006)
Cheung, C. Y. et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360, 1831–1837 (2002)
Zhou, J. et al. Functional tumor necrosis factor-related apoptosis-inducing ligand production by avian influenza virus-infected macrophages. J. Infect. Dis. 193, 945–953 (2006)
Chen, W. et al. A novel influenza A virus mitochondrial protein that induces cell death. Nature Med. 7, 1306–1312 (2001)
Zamarin, D., Garcia-Sastre, A., Xiao, X., Wang, R. & Palese, P. Influenza virus PB1–F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1, e4 (2005)
Taubenberger, J. K. et al. Characterization of the 1918 influenza virus polymerase genes. Nature 437, 889–893 (2005)
Salomon, R. et al. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J. Exp. Med. 203, 689–697 (2006)
Gabriel, G. et al. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl Acad. Sci. USA 102, 18590–18595 (2005)
Shinya, K. et al. PB2 amino acid at position 627 affects replicative efficiency, but not cell tropism, of Hong Kong H5N1 influenza A viruses in mice. Virology 320, 258–266 (2004)
Hatta, M., Gao, P., Halfmann, P. & Kawaoka, Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293, 1840–1842 (2001)
Katz, J. M. et al. Molecular correlates of influenza A H5N1 virus pathogenesis in mice. J. Virol. 74, 10807–10810 (2000)
Fodor, E. et al. Rescue of influenza A virus from recombinant DNA. J. Virol. 73, 9679–9682 (1999)
Hayden, F. G. et al. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J. Clin. Invest. 101, 643–649 (1998)
Reid, A. H., Fanning, T. G., Hultin, J. V. & Taubenberger, J. K. Origin and evolution of the 1918 ‘Spanish’ influenza virus hemagglutinin gene. Proc. Natl Acad. Sci. USA 96, 1651–1656 (1999)
Reid, A. H., Fanning, T. G., Janczewski, T. A., McCall, S. & Taubenberger, J. K. Characterization of the 1918 ‘Spanish’ influenza virus matrix gene segment. J. Virol. 76, 10717–10723 (2002)
Reid, A. H., Fanning, T. G., Janczewski, T. A. & Taubenberger, J. K. Characterization of the 1918 ‘Spanish’ influenza virus neuraminidase gene. Proc. Natl Acad. Sci. USA 97, 6785–6790 (2000)
Basler, C. F. et al. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl Acad. Sci. USA 98, 2746–2751 (2001)
Reid, A. H., Fanning, T. G., Janczewski, T. A., Lourens, R. M. & Taubenberger, J. K. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene. J. Virol. 78, 12462–12470 (2004)
Kash, J. C. et al. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 80, 3009–3020 (2006)
Brazma, A. et al. Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nature Genet. 29, 365–371 (2001)
Acknowledgements
We thank K. A. Walters, M. J. Korth, J. Fornek and B. Paeper for discussions and assistance in manuscript preparation. This work was partly supported by grants from the National Institutes of Health (to A.G.S., P.P., J.K.T. and M.G.K.) and by the Ellison Medical Foundation (P.P. and C.F.B).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Raw data on expression microarrays are available at http://expression.microslu.washington.edu. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
Supplementary information
Supplementary Figures
This file contains Supplementary Figures 1–17 and their legends. (PDF 3412 kb)
Rights and permissions
About this article
Cite this article
Kash, J., Tumpey, T., Proll, S. et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443, 578–581 (2006). https://doi.org/10.1038/nature05181
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature05181
This article is cited by
-
Aggregation-induced emission: recent applications in infectious diseases
Science China Chemistry (2023)
-
An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates
Virus Genes (2022)
-
Quantity of IgG response to SARS-CoV-2 spike glycoprotein predicts pulmonary recovery from COVID-19
Scientific Reports (2022)
-
TNF controls a speed-accuracy tradeoff in the cell death decision to restrict viral spread
Nature Communications (2021)
-
From threat to cure: understanding of virus-induced cell death leads to highly immunogenic oncolytic influenza viruses
Cell Death Discovery (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
