
Abstract
Since the discovery of the hepatitis C virus over 15 years ago, scientists have raced to develop diagnostics, study the virus and find new therapies. Yet virtually every attempt to dissect this pathogen has met with roadblocks that impeded progress. Its replication was restricted to humans or experimentally infected chimpanzees, and efficient growth of the virus in cell culture failed until very recently. Nevertheless hard-fought progress has been made and the first wave of antiviral drugs is entering clinical trials.
Similar content being viewed by others

Main
In the mid-1970s, it was noticed that the world's supply of blood was contaminated with an unidentified agent causing post-transfusion non-A, non-B hepatitis1. Yet it was not until 1989 that the first sequences of hepatitis C virus (HCV) were reported2. The difficulty in identifying the virus and in detecting viral RNA and antigens in infected tissues left the impression that HCV replicated poorly in vivo. This is not so. Chronically infected patients have viral loads that typically range from 103–107 genomes per ml of serum. Mathematical modelling of viral dynamics during treatment with interferon-α (IFN-α) indicates that HCV virions turn over rapidly (with a half-life about 3 h), and up to about 1012 viruses are produced per day in an infected person3. This is about 100-fold greater than the rate reported for HIV.
High viral loads are observed during the first few weeks of HCV infection, but inflammatory processes leading to liver injury are delayed, usually occurring after 2–3 months4. Liver transplant recipients generally have favourable short-term outcomes despite efficient allograft reinfection and high levels of viraemia owing to immunosuppression. These observations have led to the idea that HCV is relatively noncytopathic and that liver disease is immune-mediated. Although the liver is the major site of HCV replication, evidence exists for extrahepatic reservoirs including peripheral blood lymphocytes (reviewed in ref. 5), epithelial cells in the gut6 and the central nervous system7. With the hepatitis B virus immunohistochemistry can be used to identify infected hepatocytes reliably, but we do not have a clear picture of the number of HCV-infected hepatocytes in the liver or the characteristics of an HCV-infected cell. Nevertheless, gene-profiling studies of HCV-infected livers indicate that this organ is a veritable battleground of ongoing viral replication and host antiviral defences8,9,10,11. Although the origin of HCV and the timing of its introduction into the human population are not known, the high error rate of RNA-dependent RNA replication and the battle between virus and host have generated remarkable global diversity. HCV is currently divided into six major genotypes with numerous subtypes and exists as a quasispecies swarm within the infected individual12.
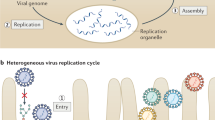
We will use an idealized HCV life cycle (Fig. 1) as a framework for discussing the current state of our knowledge. Enveloped virus particles interact with specific surface receptors and are probably internalized. Fusion of the viral and cellular membranes, presumably triggered by the low pH of the endocytic compartment, leads to the release of a single-stranded (ss), positive-sense RNA genome into the cytoplasm of a newly infected cell. This genome serves multiple roles within the virus life cycle: first, as a messenger RNA (mRNA) for translation of the viral proteins; second as a template for RNA replication; and third, as a nascent genome packaged within new virus particles. Virions presumably form by budding into the endoplasmic reticulum (ER) and leave the cell through the secretory pathway.

After entry into the cell and uncoating, the HCV genome functions in three main roles: translation, replication and packaging into nascent virions.
Researchers have followed each aspect of the virus life cycle in turn. Just as infection starts from an HCV genome entering the cytoplasm and progressing through translation, replication and particle production, our understanding has progressed from having a genome sequence to understanding translation and the viral gene products, characterizing RNA replication, and establishing systems to characterize virus particles and infectivity. In this review, we summarize the current understanding of HCV replication with special emphasis on recent developments. As space is limited, we cannot be comprehensive; readers may wish to consult other reviews for detail and breadth5,13,14.
Initial studies: HCV translation and polyprotein processing
The identification of HCV yielded a partial viral genome sequence5. Research in the early 1990s focused on dissecting HCV gene expression and characterizing the gene products. Much has been learned about the biochemistry of three key enzymes, and structural information is now available at the atomic level for roughly half of the protein-coding region.
Translation of the HCV genome, which lacks a 5′ cap, depends on an internal ribosome entry site (IRES) within the 5′-noncoding region (NCR). The HCV IRES binds 40S ribosomal subunits directly and avidly, bypassing the need for pre-initiation factors, and inducing an mRNA-bound conformation in the 40S subunit15. The IRES–40S complex then recruits eukaryotic initiation factor (eIF) 3 and the ternary complex of Met-tRNA–eIF2–GTP to form a non-canonical 48S intermediate, before a kinetically slow transition to the translationally active 80S complex16,17.
Once initiated, translation of the HCV genome produces a large polyprotein that is proteolytically cleaved to produce 10 viral proteins (Fig. 2a). The amino-terminal one-third of the polyprotein encodes the virion structural proteins: the highly basic core (C) protein, and glycoproteins E1 and E2. After the structural region comes a small integral membrane protein, p7, which seems to function as an ion channel18,19. The remainder of the genome encodes the nonstructural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A and NS5B, which coordinate the intracellular processes of the virus life cycle. The structural proteins mature by signal peptidase cleavages between C/E1, E1/E2 and E2/p7. In addition, signal-peptide peptidase releases core from the E1 signal peptide. Within the NS region, the p7/NS2 junction is also cleaved by signal peptidase. Further proteolytic processing within the NS region occurs through the action of two viral enzymes, the NS2 autoprotease, which cleaves at the NS2/3 junction; and the NS3-4A serine protease, which cleaves at all downstream sites (Fig. 2a, b). HCV also encodes a small protein, called F (frame shift) or ARFP (alternative reading frame protein), that can be produced by ribosomal frame shifting into an alternative reading frame within the core gene (reviewed in ref. 20).

a, The structure of the viral genome, including the long open reading frame encoding structural and nonstructural genes, and 5′ and 3′ NCRs. The polyprotein processing scheme is shown below. Closed circles refer to signal peptidase cleavage sites; the open circle refers to the signal peptide peptidase cleavage site. All other terms are defined in the text. b, The topology of HCV proteins with respect to a cellular membrane.
HCV encodes two remarkable proteases
The carboxy-terminal two-thirds of NS2 contain the catalytic triad of a cysteine protease. NS2/3 cleavage requires these residues as well as the downstream expression of the NS3 serine protease domain, although NS3-4A protease activity is dispensable for NS2/3 processing. The requirement for NS3 may have to do with correct folding, as NS2/3 cleavage is enhanced by Zn2+, which has a structural role in stabilizing the NS3 fold.
NS3 is a multifunctional protein, with an N-terminal serine protease domain and a C-terminal RNA helicase/NTPase domain. Both enzyme activities have been well characterized, and high-resolution structures have been solved14. The serine protease domain has a typical chymotrypsin-like fold, positioning three catalytically active residues at the surface interface between two β-barrel domains (Fig. 3a). For complete folding and serine protease activity NS3 requires the intercalation of a β-strand present in NS4A (refs 21–23). NS4A is a small (54-amino-acid) protein that anchors NS3 to cellular membranes through an N-terminal hydrophobic peptide24. Proper folding of the serine protease domain also requires coordination of Zn2+ by three cysteine residues distal from the active site. NS3 has an unusually shallow substrate-binding pocket for a serine protease, which has challenged efforts to obtain specific inhibitors. Nevertheless, the discovery of product inhibition led to the development of potent inhibitors that block NS3 serine protease activity, NS protein processing, and HCV RNA replication. For further details, see the accompanying article by De Francesco and Migliaccio (page 953).

a, NS3. The serine protease, NS4A cofactor and RNA helicase domains are shown in pink, green and blue, respectively. The serine protease and RNA helicase active site residues are indicated in red. b, NS5A domain I. Shown is a dimer, as seen in the crystal structure. Individual subunits are shown in blue and green, with their C termini (that is, leading into domain II) pointing upwards. The N termini, which presumably face the membrane, are at the bottom. The purple spheres represent Zn2+ ions. Disulphide bonds are indicated in red. Brackets indicate highly conserved surfaces. A basic groove, which may bind RNA, is also indicated. c, NS5B. Shown is the typical ‘right hand’ model of the RdRP, with palm, fingers and thumb domains in pink, blue and green, respectively. The C-terminal region, which is not part of the RdRP, is shown in yellow. Note the extensive interactions between the finger and thumb domains. In addition, a β-hairpin is shown in purple, and active site residues Asp 220 and Asp 318 are shown in red.
Dissecting the structure and function of HCV NS proteins
The C terminus of NS3 encodes a DExH/D-box RNA helicase. These enzymes use the energy of NTP hydrolysis to unwind double-stranded RNA, and NS3 unwinds RNA and DNA homoduplexes and heteroduplexes in a 3′ to 5′ direction25. The helicase mechanism is not yet fully understood, but recent kinetic analyses show that the NS3 helicase behaves like a ratcheting two-stroke motor26 and seems to function as a dimer that incrementally rips apart 18-base-pair stretches of substrate RNA27. In addition, NS3 helicase activity can be regulated by interactions between the serine protease and helicase domains of NS3 (refs 28, 29), indicating that these two enzyme activities may be somehow coordinated during replication. The function of the HCV helicase is not known, but it may be involved in the initiation of RNA synthesis on the HCV genome RNA, which contains stable 3′-terminal secondary structure, in dissociation of nascent RNA strands from their template during RNA synthesis, or in displacement of proteins or other trans-acting factors from the RNA genome.
NS5A is a fascinating protein. Many cellular proteins interact with NS5A, although their functional relevance is largely unclear30,31,32. NS5A is phosphorylated on multiple serine residues by cellular kinases and can be found in hypophosphorylated (56 kDa) and hyperphosphorylated (58 kDa) forms. Major phosphorylation sites have been determined for a few HCV isolates33,34, and kinases capable of phosphorylating NS5A have been identified. These include AKT, p70S6K, MEK1, MKK6, cAMP-dependent protein kinase A-α and casein kinase II35,36,37,38. It is not yet clear which kinases are involved in generating the different phosphoforms of NS5A, nor which phos-phorylation sites are functionally relevant, and the role of NS5A phosphorylation remains an area of intense interest.
NS5A associates with membranes through an N-terminal amphipathic α-helix39 and contains three distinct structural domains40. Biochemical and structural analyses revealed that domain I (residues 1–213) forms a dimer with a novel fold and coordinates Zn2+ through a unique motif40,41. This architecture reveals conserved external surfaces that might interact with other proteins, as well as a highly basic channel that might be involved in RNA binding (Fig. 3b). In addition, a disulphide bond was discovered between cysteine residues 142 and 190, which is a rare modification for cytosolic proteins and could represent another form of regulation. Unfortunately, structural information is not yet available for domains II and III, which contain determinants influencing the efficiency of RNA replication (see below) and NS5A phosphorylation. The C-terminal domain is not well conserved and seems quite flexible42,43.
The workhorse of the HCV RNA replication machinery is NS5B, which encodes the RNA-dependent RNA polymerase (RdRP). Primer-dependent and de novo (unprimed) initiation of RNA synthesis have been demonstrated for this protein. On the basis of the replication strategy of other positive-strand RNA viruses, HCV RNA replication probably involves de novo initiation by a multiprotein complex (replicase). NS5B has a typical ‘right hand’ polymerase structure, with catalytic sites in the base of the palm domain, surrounded by thumb and finger domains14. These latter domains fully encircle the active site, creating a channel for binding to a ssRNA template (Fig. 3c). In addition, a β-hairpin structure protrudes from the thumb toward the active site and is likely to be involved in correct positioning of the template44. The overall structure of NS5B is remarkably similar to the RdRP of bacteriophage φ6 (ref. 45), and cocrystallization of these enzymes with model substrates and nucleoside triphosphates has yielded a credible model for de novo initiation (reviewed in ref. 46). NS5B also has a low-affinity GTP-binding site, distal from the active site, which is thought to be an allosteric regulator of the finger–thumb interaction47. NS5B is tethered to membranes by a C-terminal peptide anchor48 and interacts with itself to form higher-order RdRP complexes that may have functional relevance to the membrane-bound replicase, described below49.
The replication era: reverse genetic systems for HCV
Once the highly conserved 3′ terminus of HCV was discovered and the genome sequence was completed50,51, reverse genetics with HCV became possible. Functional complementary DNA clones were assembled and shown to be infectious by direct intrahepatic injection of RNA transcripts into chimpanzees52,53. These infectious clones were used to show that all viral enzyme activities, the p7 gene and the correct genomic 3′ end are necessary for HCV replication in vivo54,55,56. In contrast, the hypervariable N-terminal region of E2 is dispensible57. Essentially clonal infections could be initiated from transcribed RNA, providing a well-defined genetic starting point to study virus evolution and immune responses to infection58. With functional cDNA clones in hand and the ability to make unlimited quantities of infectious HCV RNA, much effort was devoted to searching for permissive HCV cell culture conditions. However, despite their great utility for in vivo studies, these initial chimpanzee infectious transcripts failed to replicate in cell culture.
A breakthrough for the field came in 1999 when Lohmann et al. reported selection of the first functional ‘subgenomic’ replicons in cell culture59. These replicons consisted of a genotype 1b HCV RNA engineered to express a selectable marker gene, Neo, in place of the structural protein coding region, which was not expected to be required for RNA replication. To direct expression of NS proteins, a heterologous viral IRES was inserted after the neomycin resistance cassette (Fig. 4). After RNA transfection into a human hepatoma line, Huh-7, a few drug-selected colonies grew out that contained replicating HCV RNAs. The replicon system provided an important tool for studying HCV RNA replication and established a functional cell-based system for evaluating potential antiviral compounds.

The basic design of subgenomic replicons.
The next discovery was that replicon RNAs harboured culture-adaptive mutations, often in NS5A, that increased RNA replication and Neo-transduction efficiency by up to 10,000-fold60. Adaptive changes were subsequently mapped throughout the NS region, including NS3, NS4B, NS5A and NS5B (reviewed in ref. 13). In addition to the original genotype 1b isolate, called Con1, replicons have now been established for other 1b isolates and for genotypes 1a and 2a, and HCV RNA replication has been achieved in Hela, 293, HepG2 and even mouse hepatoma cell lines13. Thus, whereas it was once thought that HCV replication might be restricted to the environment provided by hepatocytes, it is now clear that a number of cells can support RNA replication. As discussed by Gale and Foy (see page 939, in this issue) the interplay between HCV and innate cellular antiviral pathways may be a major intracellular determinant of HCV tropism.
Mechanisms of HCV RNA replication
Characterization of cell-culture-adaptive mutations is an area of great interest, as they are likely to teach us about the interface between HCV replication and the host cellular environment. Adaptive mutations in NS4B, NS5A or NS5B strongly enhance replication but are incompatible with each other, whereas adaptive changes in NS3 tend to be weak and cooperatively enhance replication when combined with strongly adaptive mutations13. Furthermore, adaptive mutations in NS3 and NS5B map to surface residues distant from the enzyme active sites, suggesting that these changes are likely to affect interactions between NS proteins and/or cellular factors. A striking feature of highly adaptive mutations is their tendency to decrease NS5A hyperphosphorylation, and many adaptive changes map to NS5A residues implicated in this process60,61,62. Similarly, increased hyperphosphorylation of NS5A correlates with lower replication levels42,63,64 and decreased interaction between NS5A and a cellular factor, human vesicle-associated membrane-protein associated protein A (hVAP-A)63. This vesicle-sorting protein has also been shown to localize HCV NS proteins in cholesterol-rich, detergent-resistant membranes that may be subcellular microenvironments for HCV RNA replication65.
As for all positive-strand RNA viruses, HCV RNA replication occurs in association with altered cytoplasmic membranes. In replicon-bearing cells, HCV NS proteins, RNA and RdRP activity associate with ultrastructural vesicular structures termed the ‘membranous web’, which also resembles the membrane alterations seen in hepatocytes from HCV-infected liver66,67. The integral membrane protein NS4B is sufficient to induce membranous web formation and has been proposed to serve as a scaffold for replication complex assembly66. As mentioned, several adaptive mutations have been mapped to this protein, and NS4B has been found to encode a GTPase activity that may be related to its membrane-altering ability68. Association of the HCV replicase with the membranous web can be followed in live cells by using subgenomic replicons with green fluorescent protein inserted in the NS5A domain III43.
Recent experiments have shed new light on the interplay between cellular membranes, lipid metabolism and HCV replication. In cell culture, HCV RNA replication is stimulated by increasing the availability of saturated and monounsaturated fatty acids, and inhibited by polyunsaturated fatty acids or inhibitors of fatty acid synthesis69. These results suggest that membrane fluidity is important for the function of the membranous web. In addition, two groups have shown that inhibition of protein geranylgeranylation leads to the disassembly of HCV replication complexes and strong inhibition of HCV RNA replication69,70. In the absence of appropriate prenylation motifs in HCV proteins, these results suggest that a geranylgeranylated cellular protein participates in HCV replication and could be amenable to pharmacological manipulation. Recently, the geranylgeranylated host protein FBL-2 was shown to interact with NS5A and to be required for HCV RNA replication71. It is intriguing that FBL-2, which contains an ‘F-box’ motif, is likely to be involved in targeting proteins for degradation, although the identity of a relevant substrate is currently unknown.
An exciting area of progress has been the delineation of cis-acting RNA elements that guide viral replication. Nearly the entire 5′ NCR is needed for efficient RNA amplification, although a minimal replication element exists within the first 120 nucleotides (refs 72–75). As this overlaps with the IRES, there is considerable interest in understanding how this region might modulate translation and replication, which are unlikely to occur simultaneously on the same RNA template. The 3′ NCR has been found to contain a nonessential variable region, a poly-U/UC tract that must be more than 26 nucleotides long, followed by a highly conserved and essential 3′X domain55,57,76. Recently a conserved stem-loop structure within the NS5B coding region, 5BSL3.2, was found to be required for RNA replication78. Further studies indicated that 5BSL3.2 forms functionally important long-distance base pairs with the 3′X domain79. The trans-acting factors and the replication steps requiring this ‘kissing’ interaction remain to be determined.
Little is known about the process of HCV RNA synthesis within the replication complex. By inference from related viruses, RNA synthesis is likely to be semiconservative and asymmetric: the positive-strand genome serves as a template to make a negative-strand intermediate; the negative strand then serves as a template to produce multiple nascent genomes. RdRP activity can be detected in extracts from replicon-bearing cells, although this seems to reflect elongation of co-extracted templates rather than de novo initiation. Nascent products from these reactions are protected from nuclease digestion by a detergent- and protease-sensitive factor80,81. Interestingly, protease treatment of permeabilized cells destroyed most NS proteins without compromising RdRP activity, suggesting that only a small fraction of NS proteins is actively engaged in RNA replication80. The function of the ‘excess’ NS proteins, the composition of the HCV replicase and the possible reasons for physically sequestering the replicase (such as evasion of dsRNA sensors or antiviral effectors like RNAi) remain intriguing areas for future study.
Completing the virus life cycle: extracellular virions
Important questions about the nature of the infectious virus particle, the pathway of virus entry, and the assembly of viral structural proteins and RNA into new virus particles are still largely unanswered. Fortunately, new technologies have emerged that extend our reach into these formerly intractable areas.
HCV particles present in clinical samples have been partly characterized. Enveloped virions are sensitive to detergent and to chloroform, with a diameter of about 50 nm. HCV RNA and infectivity in chimpanzees have been followed in isopycnic density gradients. HCV exhibits unusual heterogeneity in buoyant density, with the peak of infectivity near 1.10 g ml−1. This is surprisingly low even for an enveloped virus. This heterogeneity and low density have been explained in part by the association of HCV particles with serum components such as immunoglobulins and β-lipoproteins5,13.
Expression and processing of the HCV structural gene products were characterized in early heterologous expression studies. Core protein was localized to the cytoplasmic surface of the ER and lipid droplets, and occasionally the cell nucleus82,83,84. A central hydrophobic domain is responsible for the membrane association of the core, whereas the high pI of an N-terminal region mediates its interaction with RNA. The envelope glycoproteins E1 and E2 are highly modified with N-linked sugars, contain intramolecular disulphide bonds and undergo a complex folding pathway that involves several ER-resident chaperones (reviewed in ref. 13). When coexpressed, E1 and E2 are retained in the ER and form non-covalent heterodimers through determinants in their transmembrane domains.
E2 binds with high affinity to the large extracellular loop of CD81, a tetraspanin that is expressed on a variety of cell types, including hepatocytes85. Although this pattern of expression does not explain the hepatotropism of HCV, CD81 is very likely to be involved in mediating HCV entry. Several other candidate HCV receptors have also been identified, including low-density lipoprotein receptor, scavenger receptor class-B type-I (SR-BI), L-SIGN and DC-SIGN13. Several surrogate systems have been developed to examine the relevance of these interactions and to study E1/E2 structure and function. These include glycoprotein-dependent cell fusion assays, liposomes reconstituted with E1 and E2, formation of virus-like particles in insect cells, and pseudotyped rhabdoviruses and retroviruses (reviewed in ref. 13). Although each of these systems had merit, the use of retrovirus pseudoparticles (HCVpp) has provided the most insight into HCV entry. This method takes advantage of the fact that retroviruses, which bud from the plasma membrane, frequently and nonspecifically incorporate cell surface proteins into the viral membrane. Thus HCVpp are retroviral particles that are dependent on HCV glycoproteins to deliver a reporter gene encoded within the retrovirus genome.
HCVpp can be neutralized with antibodies against E2 or immune sera, confirming their dependence on E2 and showing their use in following the kinetics and specificity of the humoral immune response86,87. HCVpp infect primary human hepatocytes and a variety of human hepatic cell lines, and their entry is CD81 dependent88,89,90,91. CD81 expression is not sufficient for HCV entry into non-hepatic cells, suggesting the existence of one or more unidentified molecules required for HCV entry and hepatotropism. High-density lipoproteins (HDL) and apolipoprotein C1, a component of HDL, enhance HCVpp infectivity in an SR-BI-dependent manner87,92. It is not yet clear whether these observations are related to the association of HCV with serum lipoproteins.
Despite these advances, the growth of authentic HCV in cell culture has remained elusive. Although full-length genomes harbouring adaptive mutations replicated efficiently in Huh-7 cells and expressed the structural proteins, infectious particles were not released61,93,94. This led to the idea that Huh-7 cells might be unable to support HCV particle assembly or release. However, culture-adaptive changes were also found to be lethal or highly attenuating for replication in chimpanzees95. This suggested that adaptive mutations promoting efficient RNA replication in cell culture might preclude production of infectious particles. In support of this idea, Pietschmann et al. found that full-length genomes lacking adaptive mutations replicated poorly but nevertheless released core protein and HCV RNA into the cell culture medium96. In contrast, genomes with highly adaptive mutations replicated efficiently but failed to secrete HCV RNA and core protein. Thus, there was a dilemma: in cell culture, adaptive mutations were required for replication but were deleterious for virus production; in vivo, cell adaptive mutations were deleterious and virus produced in vivo was non-infectious in cell culture. Enter JFH-1: a genotype 2a HCV isolate obtained from a patient in Japan with fulminant hepatitis97. For reasons that are not understood, subgenomic replicons derived from JFH-1 cDNA do not require adaptive mutations for efficient replication in cell culture. Wakita et al. recently demonstrated that the full-length JFH-1 genome produces infectious particles in cell culture, although the titres were moderate (ref. 98).
Similarly, we constructed a chimaeric full-length genome using the JFH-1 replicase and the core–NS2 region from a related genotype 2a stain, J6. This genome replicated in cell culture and produced robust levels of infectious virus (HCVcc), nearly 105 infectious units ml−1 within 48 h in Huh-7.5 cells, which are highly permissive for HCV RNA replication99. Another group has found that full-length JFH-1 can also reach high titres when propagated in an Huh-7.5 subline100. Thus, the JFH-1 replicon can support efficient production of infectious HCV in cell culture. It is not yet clear why this particular genome is capable of replicating without adaptive changes, or how adaptive changes preclude infectious particle production. However, one possible explanation stems from the three coordinated yet distinct processes facing HCV viral RNA: translation, replication and packaging. Cell culture adaptive changes, selecting for efficient and persistent RNA replication, may shift the balance towards these first two processes at the expense of liberating genome RNA for virion assembly.
Outlook
It is clearly an exciting time in HCV research, and rapid progress should be made now that complete cell culture systems are available. The processes of HCV entry, replication and virion production can be further dissected with genetic and biochemical approaches, and should reveal information that facilitates the development of specific antivirals that target each stage in the virus life cycle. If the determinants of JFH-1 that permit efficient replication and virus production can be mapped, it may be possible to extend the cell culture systems to include other virus isolates as well. An important question is whether JFH-1-derived viruses grown in vivo will retain their infectivity in cell culture. Furthermore, once cellular determinants of HCV tropism are better understood, it might be possible to engineer improved small-animal models of HCV infection and pathogenesis. These approaches will undoubtedly reveal new and surprising aspects of HCV replication, and arm us with better strategies to combat HCV infection and eradicate HCV-associated disease.
References
Feinstone, S. M., Kapikian, A. Z., Purcell, R. H., Alter, H. J. & Holland, P. V. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N. Engl. J. Med. 292, 767–770 (1975).
Choo, Q. -L. et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244, 359–362 (1989).
Neumann, A. U. et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282, 103–107 (1998).
Hoofnagle, J. H. Course and outcome of hepatitis C. Hepatology 36, S21–S29 (2002).
Lindenbach, B. D. & Rice, C. M. in Fields Virology (eds Knipe, D. M. & Howley, P. M.) 991–1041 (Lippincott-Raven, Philadelphia, 2001).
Deforges, S. et al. Expression of hepatitis C virus proteins in epithelial intestinal cells in vivo. J. Gen. Virol. 85, 2515–2523 (2004).
Forton, D. M., Karayiannis, P., Mahmud, N., Taylor-Robinson, S. D. & Thomas, H. C. Identification of unique hepatitis C virus quasispecies in the central nervous system and comparative analysis of internal translational efficiency of brain, liver, and serum variants. J. Virol. 78, 5170–5183 (2004).
Bigger, C. B., Brasky, K. M. & Lanford, R. E. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 75, 7059–7066 (2001).
Bigger, C. B. et al. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 78, 13779–13792 (2004).
Su, A. I. et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl Acad. Sci. USA 99, 15669–15674 (2002).
Thimme, R. et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl Acad. Sci. USA 99, 15661–15668 (2002).
Simmonds, P. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 85, 3173–3188 (2004).
Bartenschlager, R., Frese, M. & Pietschmann, T. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63, 71–180 (2004).
Penin, F., Dubuisson, J., Rey, F. A., Moradpour, D. & Pawlotsky, J. M. Structural biology of hepatitis C virus. Hepatology 39, 5–19 (2004).
Spahn, C. M. et al. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40S ribosomal subunit. Science 291, 1959–1962 (2001).
Ji, H., Fraser, C. S., Yu, Y., Leary, J. & Doudna, J. A. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc. Natl Acad. Sci. USA 101, 16990–16995 (2004).
Otto, G. A. & Puglisi, J. D. The pathway of HCV IRES-mediated translation initiation. Cell 119, 369–380 (2004).
Griffin, S. D. et al. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 535, 34–38 (2003).
Pavlovic, D. et al. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc. Natl Acad. Sci. USA 100, 6104–6108 (2003).
Branch, A. D., Stump, D. D., Gutierrez, J. A., Eng, F. & Walewski, J. L. The hepatitis C virus alternate reading frame (ARF) and its family of novel products: the alternate reading frame protein/F-protein, the double-frameshift protein, and others. Semin. Liver Dis. 25, 105–117 (2005).
Failla, C., Tomei, L. & De Francesco, R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 68, 3753–3760 (1994).
Bartenschlager, R., Ahlborn-Laake, L., Mous, J. & Jacobsen, H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J. Virol. 68, 5045–5055 (1994).
Lin, C., Thomson, J. A. & Rice, C. M. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3–NS4A serine proteinase complex in vivo and in vitro. J. Virol. 69, 4373–4380 (1995).
Wölk, B. et al. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3–NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74, 2293–2304 (2000).
Tai, C. -L., Chi, W. -K., Chen, D. -S. & Hwang, L. -H. The helicase activity associated with hepatitis C virus nonstructural protein 3 (NS3). J. Virol. 70, 8477–8484 (1996).
Levin, M. K., Gurjar, M. & Patel, S. S. A Brownian motor mechanism of translocation and strand separation by hepatitis C virus helicase. Nature Struct. Mol. Biol. 12, 429–435 (2005).
Serebrov, V. & Pyle, A. M. Periodic cycles of RNA unwinding and pausing by hepatitis C virus NS3 helicase. Nature 430, 476–480 (2004).
Frick, D. N., Rypma, R. S., Lam, A. M. & Gu, B. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J. Biol. Chem. 279, 1269–1280 (2004).
Pang, P. S., Jankowsky, E., Planet, P. J. & Pyle, A. M. The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J. 21, 1168–1176 (2002).
He, Y. & Katze, M. G. To interfere and to anti-interfere: the interplay between hepatitis C virus and interferon. Viral Immunol. 15, 95–119 (2002).
Macdonald, A. & Harris, M. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol. 85, 2485–2502 (2004).
Tellinghuisen, T. L. & Rice, C. M. Interaction between hepatitis C virus proteins and host cell factors. Curr. Opin. Microbiol. 5, 419–427 (2002).
Katze, M. G. et al. Ser2194 is a highly conserved major phosphorylation site of the hepatitis C virus nonstructural protein NS5A. Virology 278, 501–513 (2000).
Reed, K. E. & Rice, C. M. Identification of the major phosphorylation site of the hepatitis C virus H strain NS5A protein as serine 2321. J. Biol. Chem. 274, 28011–28018 (1999).
Coito, C., Diamond, D. L., Neddermann, P., Korth, M. J. & Katze, M. G. High-throughput screening of the yeast kinome: identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J. Virol. 78, 3502–3513 (2004).
Ide, Y., Tanimoto, A., Sasaguri, Y. & Padmanabhan, R. Hepatitis C virus NS5A protein is phosphorylated in vitro by a stably bound protein kinase from HeLa cells and by cAMP-dependent protein kinase A-alpha catalytic subunit. Gene 201, 151–158 (1997).
Kim, J., Lee, D. & Choe, J. Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem. Biophys. Res. Commun. 257, 777–781 (1999).
Reed, K. E., Xu, J. & Rice, C. M. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J. Virol. 71, 7187–7197 (1997).
Brass, V. et al. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277, 8130–8139 (2002).
Tellinghuisen, T. L., Marcotrigiano, J., Gorbalenya, A. E. & Rice, C. M. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279, 48576–48587 (2004).
Tellinghuisen, T. L., Marcotrigiano, J. & Rice, C. M. Structure of the zinc-binding domain of an essential replicase component of hepatitis C virus. Nature 435, 374–379 (2005).
Appel, N., Pietschmann, T. & Bartenschlager, R. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79, 3187–3194 (2005).
Moradpour, D. et al. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 78, 7400–7409 (2004).
Hong, Z. et al. A novel mechanism to ensure terminal initiation by hepatitis C virus NS5B polymerase. Virology 285, 6–11 (2001).
Butcher, S. J., Grimes, J. M., Makeyev, E. V., Bamford, D. H. & Stuart, D. I. A mechanism for initiating RNA-dependent RNA polymerization. Nature 410, 235–240 (2001).
van Dijk, A. A., Makeyev, E. V. & Bamford, D. H. Initiation of viral RNA-dependent RNA polymerization. J. Gen. Virol. 85, 1077–1093 (2004).
Bressanelli, S., Tomei, L., Rey, F. A. & De Francesco, R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 76, 3482–3492 (2002).
Schmidt-Mende, J. et al. Determinants for membrane association of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 276, 44052–44063 (2001).
Wang, Q. M. et al. Oligomerization and cooperative RNA synthesis activity of hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 76, 3865–3872 (2002).
Kolykhalov, A. A., Feinstone, S. M. & Rice, C. M. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 70, 3363–3371 (1996).
Tanaka, T., Kato, N., Cho, M. J. & Shimotohno, K. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun. 215, 744–749 (1995).
Kolykhalov, A. A. et al. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277, 570–574 (1997).
Yanagi, M., Purcell, R. H., Emerson, S. U. & Bukh, J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl Acad. Sci. USA 94, 8738–8743 (1997).
Kolykhalov, A. A., Mihalik, K., Feinstone, S. M. & Rice, C. M. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74, 2046–2051 (2000).
Sakai, A. et al. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc. Natl Acad. Sci. USA 100, 11646–11651 (2003).
Yanagi, M., St Claire, M., Emerson, S. U., Purcell, R. H. & Bukh, J. In vivo analysis of the 3′ untranslated region of the hepatitis C virus after in vitro mutagenesis of an infectious cDNA clone. Proc. Natl Acad. Sci. USA 96, 2291–2295 (1999).
Forns, X. et al. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl Acad. Sci. USA 97, 13318–13323 (2000).
Bukh, J. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 39, 1469–1475 (2004).
Lohmann, V. et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 (1999).
Blight, K. J., Kolykhalov, A. A. & Rice, C. M. Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 (2000).
Blight, K. J., McKeating, J. A., Marcotrigiano, J. & Rice, C. M. Efficient RNA replication of hepatitis C virus genotype 1a in cell culture. J. Virol. 77, 3181–3190 (2003).
Lohmann, V., Hoffmann, S., Herian, U., Penin, F. & Bartenschlager, R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77, 3007–3019 (2003).
Evans, M. J., Rice, C. M. & Goff, S. P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl Acad. Sci. USA 101, 13038–13043 (2004).
Neddermann, P. et al. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 78, 13306–13314 (2004).
Gao, L., Aizaki, H., He, J. W. & Lai, M. M. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78, 3480–3488 (2004).
Egger, D. et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76, 5974–5984 (2002).
Gosert, R. et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77, 5487–5492 (2003).
Einav, S., Elazar, M., Danieli, T. & Glenn, J. S. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J. Virol. 78, 11288–11295 (2004).
Kapadia, S. B. & Chisari, F. V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl Acad. Sci. USA 102, 2561–2566 (2005).
Ye, J. et al. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl Acad. Sci. USA 100, 15865–15870 (2003).
Wang, C. et al. Identification of FBL2 as a geranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 18, 425–434 (2005).
Friebe, P., Lohmann, V., Krieger, N. & Bartenschlager, R. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 75, 12047–12057 (2001).
Kim, Y. K., Kim, C. S., Lee, S. H. & Jang, S. K. Domains I and II in the 5′ nontranslated region of the HCV genome are required for RNA replication. Biochem. Biophys. Res. Commun. 290, 105–112 (2002).
Luo, G., Xin, S. & Cai, Z. Role of the 5′-proximal stem-loop structure of the 5′ untranslated region in replication and translation of hepatitis C virus RNA. J. Virol. 77, 3312–3318 (2003).
Reusken, C. B., Dalebout, T. J., Eerligh, P., Bredenbeek, P. J. & Spaan, W. J. Analysis of hepatitis C virus/classical swine fever virus chimeric 5′NTRs: sequences within the hepatitis C virus IRES are required for viral RNA replication. J. Gen. Virol. 84, 1761–1769 (2003).
Friebe, P. & Bartenschlager, R. Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J. Virol. 76, 5326–5338 (2002).
Yi, M. & Lemon, S. M. 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 77, 3557–3568 (2003).
You, S., Stump, D. D., Branch, A. D. & Rice, C. M. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 78, 1352–1366 (2004).
Friebe, P., Boudet, J., Simorre, J. P. & Bartenschlager, R. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 79, 380–392 (2005).
Miyanari, Y. et al. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278, 50301–50308 (2003).
Yang, G. et al. Newly synthesized hepatitis C virus replicon RNA is protected from nuclease activity by a protease-sensitive factor(s). J. Virol. 78, 10202–10205 (2004).
Barba, G. et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl Acad. Sci. USA 94, 1200–1205 (1997).
Moradpour, D., Englert, C., Wakita, T. & Wands, J. R. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology 222, 51–63 (1996).
Yasui, K. et al. The native form and maturation process of hepatitis C virus core protein. J. Virol. 72, 6048–6055 (1998).
Pileri, P. et al. Binding of hepatitis C virus to CD81. Science 282, 938–941 (1998).
Logvinoff, C. et al. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl Acad. Sci. USA 101, 10149–10154 (2004).
Meunier, J. C. et al. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl Acad. Sci. USA 102, 4560–4565 (2005).
Cormier, E. G. et al. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl Acad. Sci. USA 101, 7270–7274 (2004).
Lavillette, D. et al. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41, 265–274 (2005).
McKeating, J. A. et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 78, 8496–8505 (2004).
Zhang, J. et al. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 78, 1448–1455 (2004).
Voisset, C. et al. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 280, 7793–7799 (2005).
Blight, K. J., McKeating, J. A. & Rice, C. M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76, 13001–13014 (2002).
Pietschmann, T. et al. Persistant and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76, 4008–4021 (2002).
Bukh, J. et al. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc. Natl Acad. Sci. USA 99, 14416–14421 (2002).
Pietschmann, T. & Bartenschlager, R. in Proc. 11th International Symposium on Hepatitis C Virus and Related Viruses (Heidelberg, Germany, 2004).
Kato, T. et al. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125, 1808–1817 (2003).
Wakita, T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Med. 11, 791–796 (2005).
Lindenbach, B. D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 (2005).
Zhong, J. et al. Robust hepatitis C virus infection in vitro. Proc. Natl Acad. Sci. USA 102, 9294–9299 (2005).
Acknowledgements
We thank our colleagues for many helpful discussions, and in particular T. Tellinghuisen, J. Marcotrigiano, I. Lorenz, T. Pietschmann and R. Bartenschlager for providing data before publication; and J. Bloom, T. Tellinghuisen, M. Evans and I. Lorenz for comments on the manuscript. Work in our laboratory is supported by the US Public Health Service under grants from the NIH to C.M.R., and the Greenberg Medical Research Institute. B.D.L. is a recipient of the NIH Howard Temin Award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The infection system cited in the review and published in Science has been patented by the Rockefeller University and licenses are being sought. The Huh-7.5 cells used for HCV propagation are licensed to Apath, UC, a commercial entity in which Dr Rice is a major shareholder.
Additional information
Author Information Reprints and permissions information is available at npg.nature.com/reprintsandpermissions.
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Lindenbach, B., Rice, C. Unravelling hepatitis C virus replication from genome to function. Nature 436, 933–938 (2005). https://doi.org/10.1038/nature04077
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature04077
This article is cited by
-
Repurposing of drug molecules from FDA database against Hepatitis C virus E2 protein using ensemble docking approach
Molecular Diversity (2023)
-
Major genomic mutations driving hepatocellular carcinoma
Genome Instability & Disease (2023)
-
Target-specificity of different amyrin subunits in impeding HCV influx mechanism inside the human cells considering the quantum tunnel profiles and molecular strings of the CD81 receptor: a combined in silico and in vivo study
In Silico Pharmacology (2023)
-
NS34A resistance-associated substitutions in chronic hepatitis C in Upper Egypt and regression of liver fibrosis after direct-acting antiviral therapy
Egyptian Liver Journal (2021)
-
Adenosylhomocysteinase like 1 interacts with nonstructural 5A and regulates hepatitis C virus propagation
Journal of Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
