
Abstract
Here we present a draft genome sequence of the common chimpanzee (Pan troglodytes). Through comparison with the human genome, we have generated a largely complete catalogue of the genetic differences that have accumulated since the human and chimpanzee species diverged from our common ancestor, constituting approximately thirty-five million single-nucleotide changes, five million insertion/deletion events, and various chromosomal rearrangements. We use this catalogue to explore the magnitude and regional variation of mutational forces shaping these two genomes, and the strength of positive and negative selection acting on their genes. In particular, we find that the patterns of evolution in human and chimpanzee protein-coding genes are highly correlated and dominated by the fixation of neutral and slightly deleterious alleles. We also use the chimpanzee genome as an outgroup to investigate human population genetics and identify signatures of selective sweeps in recent human evolution.
Similar content being viewed by others
Main
More than a century ago Darwin1 and Huxley2 posited that humans share recent common ancestors with the African great apes. Modern molecular studies have spectacularly confirmed this prediction and have refined the relationships, showing that the common chimpanzee (Pan troglodytes) and bonobo (Pan paniscus or pygmy chimpanzee) are our closest living evolutionary relatives3. Chimpanzees are thus especially suited to teach us about ourselves, both in terms of their similarities and differences with human. For example, Goodall's pioneering studies on the common chimpanzee revealed startling behavioural similarities such as tool use and group aggression4,5. By contrast, other features are obviously specific to humans, including habitual bipedality, a greatly enlarged brain and complex language5. Important similarities and differences have also been noted for the incidence and severity of several major human diseases6.
Genome comparisons of human and chimpanzee can help to reveal the molecular basis for these traits as well as the evolutionary forces that have moulded our species, including underlying mutational processes and selective constraints. Early studies sought to draw inferences from sets of a few dozen genes7,8,9, whereas recent studies have examined larger data sets such as protein-coding exons10, random genomic sequences11,12 and an entire chimpanzee chromosome13.
Here we report a draft sequence of the genome of the common chimpanzee, and undertake comparative analyses with the human genome. This comparison differs fundamentally from recent comparative genomic studies of mouse, rat, chicken and fish14,15,16,17. Because these species have diverged substantially from the human lineage, the focus in such studies is on accurate alignment of the genomes and recognition of regions of unusually high evolutionary conservation to pinpoint functional elements. Because the chimpanzee lies at such a short evolutionary distance with respect to human, nearly all of the bases are identical by descent and sequences can be readily aligned except in recently derived, large repetitive regions. The focus thus turns to differences rather than similarities. An observed difference at a site nearly always represents a single event, not multiple independent changes over time. Most of the differences reflect random genetic drift, and thus they hold extensive information about mutational processes and negative selection that can be readily mined with current analytical techniques. Hidden among the differences is a minority of functionally important changes that underlie the phenotypic differences between the two species. Our ability to distinguish such sites is currently quite limited, but the catalogue of human–chimpanzee differences opens this issue to systematic investigation for the first time. We would also hope that, in elaborating the few differences that separate the two species, we will increase pressure to save chimpanzees and other great apes in the wild.
Our results confirm many earlier observations, but notably challenge some previous claims based on more limited data. The genome-wide data also allow some questions to be addressed for the first time. (Here and throughout, we refer to chimpanzee–human comparison as representing hominids and mouse–rat comparison as representing murids—of course, each pair covers only a subset of the clade.) The main findings include:
-
Single-nucleotide substitutions occur at a mean rate of 1.23% between copies of the human and chimpanzee genome, with 1.06% or less corresponding to fixed divergence between the species.
-
Regional variation in nucleotide substitution rates is conserved between the hominid and murid genomes, but rates in subtelomeric regions are disproportionately elevated in the hominids.
-
Substitutions at CpG dinucleotides, which constitute one-quarter of all observed substitutions, occur at more similar rates in male and female germ lines than non-CpG substitutions.
-
Insertion and deletion (indel) events are fewer in number than single-nucleotide substitutions, but result in ∼1.5% of the euchromatic sequence in each species being lineage-specific.
-
There are notable differences in the rate of transposable element insertions: short interspersed elements (SINEs) have been threefold more active in humans, whereas chimpanzees have acquired two new families of retroviral elements.
-
Orthologous proteins in human and chimpanzee are extremely similar, with ∼29% being identical and the typical orthologue differing by only two amino acids, one per lineage.
-
The normalized rates of amino-acid-altering substitutions in the hominid lineages are elevated relative to the murid lineages, but close to that seen for common human polymorphisms, implying that positive selection during hominid evolution accounts for a smaller fraction of protein divergence than suggested in some previous reports.
-
The substitution rate at silent sites in exons is lower than the rate at nearby intronic sites, consistent with weak purifying selection on silent sites in mammals.
-
Analysis of the pattern of human diversity relative to hominid divergence identifies several loci as potential candidates for strong selective sweeps in recent human history.
In this paper, we begin with information about the generation, assembly and evaluation of the draft genome sequence. We then explore overall genome evolution, with the aim of understanding mutational processes at work in the human genome. We next focus on the evolution of protein-coding genes, with the aim of characterizing the nature of selection. Finally, we briefly discuss initial insights into human population genetics.
In recognition of its strong community support, we will refer to chimpanzee chromosomes using the orthologous numbering nomenclature proposed by ref. 18, which renumbers the chromosomes of the great apes from the International System for Human Cytogenetic Nomenclature (ISCN; 1978) standard to directly correspond to their human orthologues, using the terms 2A and 2B for the two ape chromosomes corresponding to human chromosome 2.
Genome sequencing and assembly
We sequenced the genome of a single male chimpanzee (Clint; Yerkes pedigree number C0471; Supplementary Table S1), a captive-born descendant of chimpanzees from the West Africa subspecies Pan troglodytes verus, using a whole-genome shotgun (WGS) approach19,20. The data were assembled using both the PCAP and ARACHNE programs21,22 (see Supplementary Information ‘Genome sequencing and assembly’ and Supplementary Tables S2–S6). The former was a de novo assembly, whereas the latter made limited use of human genome sequence (NCBI build 34)23,24 to facilitate and confirm contig linking. The ARACHNE assembly has slightly greater continuity (Table 1) and was used for analysis in this paper. The draft genome assembly—generated from ∼3.6-fold sequence redundancy of the autosomes and ∼1.8-fold redundancy of both sex chromosomes—covers ∼94% of the chimpanzee genome with >98% of the sequence in high-quality bases. A total of 50% of the sequence (N50) is contained in contigs of length greater than 15.7 kilobases (kb) and supercontigs of length greater than 8.6 megabases (Mb). The assembly represents a consensus of two haplotypes, with one allele from each heterozygous position arbitrarily represented in the sequence.
Assessment of quality and coverage. The chimpanzee genome assembly was subjected to rigorous quality assessment, based on comparison to finished chimpanzee bacterial artificial chromosomes (BACs) and to the human genome (see Supplementary Information ‘Genome sequencing and assembly’ and Supplementary Tables S7–S16).
Nucleotide-level accuracy is high by several measures. About 98% of the chimpanzee genome sequence has quality scores25 of at least 40 (Q40), corresponding to an error rate of ≤10-4. Comparison of the WGS sequence to 1.3 Mb of finished BACs from the sequenced individual is consistent with this estimate, giving a high-quality discrepancy rate of 3 × 10-4 substitutions and 2 × 10-4 indels, which is no more than expected given the heterozygosity rate (see below), as 50% of the polymorphic alleles in the WGS sequence will differ from the single-haplotype BACs. Comparison of protein-coding regions aligned between the WGS sequence, the recently published sequence of chimpanzee chromosome 21 (ref. 13; formerly chromosome 22 (ref. 18)) and the human genome also revealed no excess of substitutions in the WGS sequence (see Supplementary Information ‘Genome sequencing and assembly’). Thus, by restricting our analysis to high-quality bases, the nucleotide-level accuracy of the WGS assembly is essentially equal to that of ‘finished’ sequence.
Structural accuracy is also high based on comparison with finished BACs from the primary donor and other chimpanzees, although the relatively low level of sequence redundancy limits local contiguity. On the basis of comparisons with the primary donor, some small supercontigs (most <5 kb) have not been positioned within large supercontigs (∼1 event per 100 kb); these are not strictly errors but nonetheless affect the utility of the assembly. There are also small, undetected overlaps (all <1 kb) between consecutive contigs (∼1.2 events per 100 kb) and occasional local misordering of small contigs (∼0.2 events per 100 kb). No misoriented contigs were found. Comparison with the finished chromosome 21 sequence yielded similar discrepancy rates (see Supplementary Information ‘Genome sequencing and assembly’).
The most problematic regions are those containing recent segmental duplications. Analysis of BAC clones from duplicated (n = 75) and unique (n = 28) regions showed that the former tend to be fragmented into more contigs (1.6-fold) and more supercontigs (3.2-fold). Discrepancies in contig order are also more frequent in duplicated than unique regions (∼0.4 versus ∼0.1 events per 100 kb). The rate is twofold higher in duplicated regions with the highest sequence identity (> 98%). If we restrict the analysis to older duplications (≤ 98% identity) we find fewer assembly problems: 72% of those that can be mapped to the human genome are shared as duplications in both species. These results are consistent with the described limitations of current WGS assembly for regions of segmental duplication26. Detailed analysis of these rapidly changing regions of the genome is being performed with more directed approaches27.
Chimpanzee polymorphisms
The draft sequence of the chimpanzee genome also facilitates genome-wide studies of genetic diversity among chimpanzees, extending recent work28,29,30,31. We sequenced and analysed sequence reads from the primary donor, four other West African and three central African chimpanzees (Pan troglodytes troglodytes) to discover polymorphic positions within and between these individuals (Supplementary Table S17).
A total of 1.66 million high-quality single-nucleotide polymorphisms (SNPs) were identified, of which 1.01 million are heterozygous within the primary donor, Clint. Heterozygosity rates were estimated to be 9.5 × 10-4 for Clint, 8.0 × 10-4 among West African chimpanzees and 17.6 × 10-4 among central African chimpanzees, with the variation between West and central African chimpanzees being 19.0 × 10-4. The diversity in West African chimpanzees is similar to that seen for human populations32, whereas the level for central African chimpanzees is roughly twice as high.
The observed heterozygosity in Clint is broadly consistent with West African origin, although there are a small number of regions of distinctly higher heterozygosity. These may reflect a small amount of central African ancestry, but more likely reflect undetected regions of segmental duplications present only in chimpanzees.
Genome evolution
We set out to study the mutational events that have shaped the human and chimpanzee genomes since their last common ancestor. We explored changes at the level of single nucleotides, small insertions and deletions, interspersed repeats and chromosomal rearrangements. The analysis is nearly definitive for the smallest changes, but is more limited for larger changes, particularly lineage-specific segmental duplications, owing to the draft nature of the genome sequence.
Nucleotide divergence
Best reciprocal nucleotide-level alignments of the chimpanzee and human genomes cover ∼2.4 gigabases (Gb) of high-quality sequence, including 89 Mb from chromosome X and 7.5 Mb from chromosome Y.
Genome-wide rates. We calculate the genome-wide nucleotide divergence between human and chimpanzee to be 1.23%, confirming recent results from more limited studies12,33,34. The differences between one copy of the human genome and one copy of the chimpanzee genome include both the sites of fixed divergence between the species and some polymorphic sites within each species. By correcting for the estimated coalescence times in the human and chimpanzee populations (see Supplementary Information ‘Genome evolution’), we estimate that polymorphism accounts for 14–22% of the observed divergence rate and thus that the fixed divergence is ∼1.06% or less.
Nucleotide divergence rates are not constant across the genome, as has been seen in comparisons of the human and murid genomes16,17,24,35,36. The average divergence in 1-Mb segments fluctuates with a standard deviation of 0.25% (coefficient of variation = 0.20), which is much greater than the 0.02% expected assuming a uniform divergence rate (Fig. 1a; see also Supplementary Fig. S1).

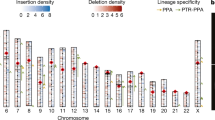
a, Distribution of divergence of the autosomes (blue), the X chromosome (red) and the Y chromosome (green). b, Distribution of variation by chromosome, shown as a box plot. The edges of the box correspond to quartiles; the notches to the standard error of the median; and the vertical bars to the range. The X and Y chromosomes are clear outliers, but there is also high local variation within each of the autosomes.
Regional variation in divergence could reflect local variation in either mutation rate or other evolutionary forces. Among the latter, one important force is genetic drift, which can cause substantial differences in divergence time across loci when comparing closely related species, as the divergence time for orthologues is the sum of two terms: t1, the time since speciation, and t2, the coalescence time for orthologues within the common ancestral population37. Whereas t1 is constant across loci (∼6–7 million years38), t2 is a random variable that fluctuates across loci (with a mean that depends on population size and here may be on the order of 1–2 million years39). However, because of historical recombination, the characteristic scale of such fluctuations will be on the order of tens of kilobases, which is too small to account for the variation observed for 1-Mb regions40 (see Supplementary Information ‘Genome evolution’). Other potential evolutionary forces are positive or negative selection. Although it is more difficult to quantify the expected contributions of selection in the ancestral population41,42,43, it is clear that the effects would have to be very strong to explain the large-scale variation observed across mammalian genomes16,44. There is tentative evidence from in-depth analysis of divergence and diversity that natural selection is not the major contributor to the large-scale patterns of genetic variability in humans45,46,47. For these reasons, we suggest that the large-scale variation in the human–chimpanzee divergence rate primarily reflects regional variation in mutation rate.
Chromosomal variation in divergence rate. Variation in divergence rate is evident even at the level of whole chromosomes (Fig. 1b). The most striking outliers are the sex chromosomes, with a mean divergence of 1.9% for chromosome Y and 0.94% for chromosome X. The likely explanation is a higher mutation rate in the male compared with female germ line48. Indeed, the ratio of the male/female mutation rates (denoted α) can be estimated by comparing the divergence rates among the sex chromosomes and the autosomes and correcting for ancestral polymorphism as a function of population size of the most recent common ancestor (MRCA; see Supplementary Information ‘Genome evolution’). Estimates for α range from 3 to 6, depending on the chromosomes compared and the assumed ancestral population size (Supplementary Table S18). This is significantly higher than recent estimates of α for the murids (∼1.9) (ref. ref. 17) and resolves a recent controversy based on smaller data sets12,24,49,50.
The higher mutation rate in the male germ line is generally attributed to the 5–6-fold higher number of cell divisions undergone by male germ cells48. We reasoned that this would affect mutations resulting from DNA replication errors (the rate should scale with the number of cell divisions) but not mutations resulting from DNA damage such as deamination of methyl CpG to TpG (the rate should scale with time). Accordingly, we calculated α separately for CpG sites, obtaining a value of ∼2 from the comparison of rates between autosomes and chromosome X. This intermediate value is a composite of the rates of CpG loss and gain, and is consistent with roughly equal rates of CpG to TpG transitions in the male and female germ line51,52.
Significant variation in divergence rates is also seen among autosomes (Fig. 1b; P < 3 × 10-15, Kruskal–Wallis test over 1-Mb windows), confirming earlier observations based on low-coverage WGS sampling12. Additional factors thus influence the rate of divergence between chimpanzee and human chromosomes. These factors are likely to act at length scales significantly shorter than a chromosome, because the standard deviation across autosomes (0.21%) is comparable to the standard deviation seen in 1-Mb windows across the genome (0.13–0.35%). We therefore sought to understand local factors that contribute to variation in divergence rate.
Contribution of CpG dinucleotides. Sites containing CpG dinucleotides in either species show a substantially elevated divergence rate of 15.2% per base; they account for 25.2% of all substitutions while constituting only 2.1% of all aligned bases. The divergence at CpG sites represents both the loss of ancestral CpGs and the creation of new CpGs. The former process is known to occur at a rapid rate per base due to frequent methylation of cytosines in a CpG context and their frequent deamination53,54, whereas the latter process probably proceeds at a rate more typical of other nucleotide substitutions. Assuming that loss and creation of CpG sites are close to equilibrium, the mutation rate for bases in a CpG dinucleotide must be 10–12-fold higher than for other bases (see Supplementary Information ‘Genome evolution’ and ref. 51).
Because of the high rate of CpG substitutions, regional divergence rates would be expected to correlate with regional CpG density. CpG density indeed varies across 1-Mb windows (mean = 2.1%, coefficient of variation = 0.44 compared with 0.0093 expected under a Poisson distribution), but only explains 4% of the divergence rate variance. In fact, regional CpG and non-CpG divergence is highly correlated (r = 0.88; Supplementary Fig. S2), suggesting that higher-order effects modulate the rates of two very different mutation processes (see also ref. 47).
Increased divergence in distal regions. The most striking regional pattern is a consistent increase in divergence towards the ends of most chromosomes (Fig. 2). The terminal 10 Mb of chromosomes (including distal regions and proximal regions of acrocentric chromosomes) averages 15% higher divergence than the rest of the genome (Mann–Whitney U-test; P < 10-30), with a sharp increase towards the telomeres. The phenomenon correlates better with physical distance than relative position along the chromosomes and may partially explain why smaller chromosomes tend to have higher divergence (Supplementary Fig. S3; see also ref. 15). These observations suggest that large-scale chromosomal structure, directly or indirectly, influences regional divergence patterns. The cause of this effect is unclear, but these regions (∼15% of the genome) are notable in having high local recombination rate, high gene density and high G + C content.

Human–chimpanzee divergence (blue), G + C content (green) and human recombination rates173 (red) in sliding 1-Mb windows for human and chimpanzee chromosome 1. Divergence and G + C content are noticeably elevated near the 1p telomere, a trend that holds for most subtelomeric regions (see text). Internally on the chromosome, regions of low G + C content and high divergence often correspond to the dark G bands.
Correlation with chromosome banding. Another interesting pattern is that divergence increases with the intensity of Giemsa staining in cytogenetically defined chromosome bands, with the regions corresponding to Giemsa dark bands (G bands) showing 10% higher divergence than the genome-wide average (Mann–Whitney U-test; P < 10-14) (see Fig. 2). In contrast to terminal regions, these regions (17% of the genome) tend to be gene poor, (G + C)-poor and low in recombination55,56. The elevated divergence seen in two such different types of regions suggests that multiple mechanisms are at work, and that no single known factor, such as G + C content or recombination rate, is an adequate predictor of regional variation in the mammalian genome by itself (Fig. 3). Elucidation of the relative contributions of these and other mechanisms will be important for formulating accurate models for population genetics, natural selection, divergence times and the evolution of genome-wide sequence composition57.

Conditional on recombination rate, the relationship between divergence and G + C content varies. In regions with recombination rates less than 0.8 cM Mb-1 (blue), there is an inverse relationship, where high divergence regions tend to be (G + C)-poor and low divergence regions tend to be (G + C)-rich. In regions with recombination rates greater than 2.0 cM Mb-1, whether within 10 Mb (red) or proximal (green) of chromosome ends, both divergence and G + C content are uniformly high.
Correlation with regional variation in the murid genome. Given that sequence divergence shows regional variation in both hominids (human–chimpanzee) and murids (mouse–rat), we asked whether the regional rates are positively correlated between orthologous regions. Such a correlation would suggest that the divergence rate is driven, in part, by factors that have been conserved over the ∼75 million years since rodents, humans and apes shared a common ancestor. Comparative analysis of the human and murid genomes has suggested such a correlation58,59,60, but the chimpanzee sequence provides a direct opportunity to compare independent evolutionary processes between two mammalian clades.
We compared the local divergence rates in hominids and murids across major orthologous segments in the respective genomes (Fig. 4). For orthologous segments that are non-distal in both hominids and murids, there is a strong correlation between the divergence rates (r = 0.5, P < 10-11). In contrast, orthologous segments that are centred within 10 Mb of a hominid telomere have disproportionately high divergence rates and G + C content relative to the murids (Mann–Whitney U-test; P < 10-11 and P < 10-4), implying that the elevation in these regions is, at least partially, lineage specific. The same general effect is observed (albeit less pronounced) if CpG dinucleotides are excluded (Supplementary Fig. S4). Increased divergence and G + C content might be explained by ‘biased gene conversion’61 due to the high hominid recombination rates in these distal regions. Segments that are distal in murids do not show elevated divergence rates, which is consistent with this model, because the recombination rates of distal regions are not as elevated in mouse and rat62.

Scatter plot of the ratio of human–chimpanzee divergence over mouse–rat divergence versus the ratio of human G + C content over mouse G + C content across 199 syntenic blocks for which more than 1 Mb of sequence could be aligned between all four species. Blocks for which the centre is within 10 Mb of a telomere in hominids only (green) or in hominids and murids (magenta), but not in murids only (light blue), show a significant trend towards higher ratios than internal blocks (dark blue). Blocks on the X chromosome (red) tend to show a lower divergence ratio than autosomal blocks, consistent with a smaller difference between autosomal and X divergence in murids than in hominids (lower α).
Taken together, these observations suggest that sequence divergence rate is influenced by both conserved factors (stable across mammalian evolution) and lineage-specific factors (such as proximity to the telomere or recombination rate, which may change with chromosomal rearrangements).
Insertions and deletions
We next studied the indel events that have occurred in the human and chimpanzee lineages by aligning the genome sequences to identify length differences. We will refer below to all events as insertions relative to the other genome, although they may represent insertions or deletions relative to the genome of the common ancestor.
The observable insertions fall into two classes: (1) ‘completely covered’ insertions, occurring within continuous sequence in both species; and (2) ‘incompletely covered’ insertions, occurring within sequence containing one or more gaps in the chimpanzee, but revealed by a clear discrepancy between the species in sequence length. Different methods are needed for reliable identification of modest-sized insertions (1 base to 15 kb) and large insertions (> 15 kb), with the latter only being reliably identifiable in the human genome (see Supplementary Information ‘Genome evolution’).
The analysis of modest-sized insertions reveals ∼32 Mb of human-specific sequence and ∼35 Mb of chimpanzee-specific sequence, contained in ∼5 million events in each species (Supplementary Information ‘Genome evolution’ and Supplementary Fig. S5). Nearly all of the human insertions are completely covered, whereas only half of the chimpanzee insertions are completely covered. Analysis of the completely covered insertions shows that the vast majority are small (45% of events cover only 1 base pair (bp), 96% are <20 bp and 98.6% are <80 bp), but that the largest few contain most of the sequence (with the ∼70,000 indels larger than 80 bp comprising 73% of the affected base pairs) (Fig. 5). The latter indels >80 bp fall into three categories: (1) about one-quarter are newly inserted transposable elements; (2) more than one-third are due to microsatellite and satellite sequences; (3) and the remainder are assumed to be mostly deletions in the other genome.

Sequences present in chimpanzee but not in human (blue) or present in human but not in chimpanzee (red) are shown. The prominent spike around 300 nucleotides corresponds to SINE insertion events. Most of the indels are smaller than 20 bp, but larger indels account for the bulk of lineage-specific sequence in the two genomes.
The analysis of larger insertions (> 15 kb) identified 163 human regions containing 8.3 Mb of human-specific sequence in total (Fig. 6). These cases include 34 regions that involve exons from known genes, which are discussed in a subsequent section. Although we have no direct measure of large insertions in the chimpanzee genome, it appears likely that the situation is similar.

Both the total number of candidate human insertions/chimpanzee deletions (blue) and the number of bases altered (red) are shown.
On the basis of this analysis, we estimate that the human and chimpanzee genomes each contain 40–45 Mb of species-specific euchromatic sequence, and the indel differences between the genomes thus total ∼90 Mb. This difference corresponds to ∼3% of both genomes and dwarfs the 1.23% difference resulting from nucleotide substitutions; this confirms and extends several recent studies63,64,65,66,67. Of course, the number of indel events is far fewer than the number of substitution events (∼5 million compared with ∼35 million, respectively).
Transposable element insertions
We next used the catalogue of lineage-specific transposable element copies to compare the activity of transposons in the human and chimpanzee lineages (Table 2).
Endogenous retroviruses. Endogenous retroviruses (ERVs) have become all but extinct in the human lineage, with only a single retrovirus (human endogenous retrovirus K (HERV-K)) still active24. HERV-K was found to be active in both lineages, with at least 73 human-specific insertions (7 full length and 66 solo long terminal repeats (LTRs)) and at least 45 chimpanzee-specific insertions (1 full length and 44 solo LTRs). A few other ERV classes persisted in the human genome beyond the human–chimpanzee split, leaving ∼9 human-specific insertions (all solo LTRs, including five HERV9 elements) before dying out.
Against this background, it was surprising to find that the chimpanzee genome has two active retroviral elements (PtERV1 and PtERV2) that are unlike any older elements in either genome; these must have been introduced by infection of the chimpanzee germ line. The smaller family (PtERV2) has only a few dozen copies, which nonetheless represent multiple (∼5–8) invasions, because the sequence differences among reconstructed subfamilies are too great (∼8%) to have arisen by mutation since divergence from human. It is closely related to a baboon endogenous retrovirus (BaEV, 88% ORF2 product identity) and a feline endogenous virus (ECE-1, 86% ORF2 product identity). The larger family (PtERV1) is more homogeneous and has over 200 copies. Whereas older ERVs, like HERV-K, are primarily represented by solo LTRs resulting from LTR–LTR recombination, more than half of the PtERV1 copies are still full length, probably reflecting the young age of the elements. PtERV1-like elements are present in the rhesus monkey, olive baboon and African great apes but not in human, orang-utan or gibbon, suggesting separate germline invasions in these species68.
Higher Alu activity in humans. SINE (Alu) elements have been threefold more active in humans than chimpanzee (∼7,000 compared with ∼2,300 lineage-specific copies in the aligned portion), refining the rather broad range (2–7-fold) estimated in smaller studies13,67,69. Most chimpanzee-specific elements belong to a subfamily (AluYc1) that is very similar to the source gene in the common ancestor. By contrast, most human-specific Alu elements belong to two new subfamilies (AluYa5 and AluYb8) that have evolved since the chimpanzee–human divergence and differ substantially from the ancestral source gene69. It seems likely that the resurgence of Alu elements in humans is due to these potent new source genes. However, based on an examination of available finished sequence, the baboon shows a 1.6-fold higher Alu activity relative to human new insertions, suggesting that there may also have been a general decline in activity in the chimpanzee67.
Some of the human-specific Alu elements are highly diverged (92 with >5% divergence), which would seem to suggest that they are much older than the human–chimpanzee split. Possible explanations include: gene conversion by nearby older elements; processed pseudogenes arising from a spurious transcription of an older element; precise excision from the chimpanzee genome; or high local mutation rate. In any case, the presence of such anomalies suggests that caution is warranted in the use of single-repeat elements as homoplasy-free phylogenetic markers.
New Alu elements target (A + T)-rich DNA in human and chimpanzee genomes. Older SINE elements are preferentially found in gene-rich, (G + C)-rich regions, whereas younger SINE elements are found in gene-poor, (A + T)-rich regions where long interspersed element (LINE)-1 (L1) copies also accumulate24,70. The latter distribution is consistent with the fact that Alu retrotransposition is mediated by L1 (ref. 71). Murid genomes revealed no change in SINE distribution with age17.
The human pattern might reflect either preferential retention of SINEs in (G + C)-rich regions, due to selection or mutation bias, or a recent change in Alu insertion preferences. With the availability of the chimpanzee genome, it is possible to classify the youngest Alu copies more accurately and thus begin to distinguish these possibilities.
Analysis shows that lineage-specific SINEs in both human and chimpanzee are biased towards (A + T)-rich regions, as opposed to even the most recent copies in the MRCA (Fig. 7). This indicates that SINEs are indeed preferentially retained in (G + C)-rich DNA, but comparison with a more distant primate is required to formally rule out the possibility that the insertion bias of SINEs did not change just before speciation.

Alu elements that inserted after human–chimpanzee divergence are densest in the (G + C)-poor regions of the genome (peaking at 36–40% G + C), whereas older copies, common to both genomes, crowd (G + C)-rich regions. The figure is similar to figure 23 of ref. 24, but the use of chimpanzee allows improved separation of young and old elements, leading to a sharper transition in the pattern.
Equal activity of L1 in both species. The human and chimpanzee genomes both show ∼2,000 lineage-specific L1 elements, contrary to previous estimates based on small samples that L1 activity is 2–3-fold higher in chimpanzee72.
Transcription from L1 source genes can sometimes continue into 3′ flanking regions, which can then be co-transposed73,74. Human–chimpanzee comparison revealed that ∼15% of the species-specific insertions appear to have carried with them at least 50 bp of flanking sequence (followed by a poly(A) tail and a target site duplication). In principle, incomplete reverse transcription could result in insertions of the flanking sequence only (without any L1 sequence), mobilizing gene elements such as exons, but we found no evidence of this.
Retrotransposed gene copies. The L1 machinery also mediates retrotransposition of host messenger RNAs, resulting in many intronless (processed) pseudogenes in the human genome75,76,77. We identified 163 lineage-specific retrotransposed gene copies in human and 246 in chimpanzee (Supplementary Table S19). Correcting for incomplete sequence coverage of the chimpanzee genome, we estimate that there are ∼200 and ∼300 processed gene copies in human and chimpanzee, respectively. Processed genes thus appear to have arisen at a rate of ∼50 per million years since the divergence of human and chimpanzee; this is lower than the estimated rate for early primate evolution75, perhaps reflecting the overall decrease in L1 activity. As expected78, ribosomal protein genes constitute the largest class in both species. The second largest class in chimpanzee corresponds to zinc finger C2H2 genes, which are not a major class in the human genome.
The retrotransposon SVA and distribution of CpG islands by transposable elements. The third most active element since speciation has been SVA, which created about 1,000 copies in each lineage. SVA is a composite element (∼1.5–2.5 kb) consisting of two Alu fragments, a tandem repeat and a region apparently derived from the 3′ end of a HERV-K transcript; it is probably mobilized by L1 (refs 79, 80). This element is of particular interest because each copy carries a sequence that satisfies the definition of a CpG island81 and contains potential transcription factor binding sites; the dispersion of 1,000 SVA copies could therefore be a source of regulatory differences between chimpanzee and human (Supplementary Table S20). At least three human genes contain SVA insertions near their promoters (Supplementary Table S21), one of which has been found to be differentially expressed between the two species82,83, but additional investigations will be required to determine whether the SVA insertion directly caused this difference.
Homologous recombination between interspersed repeats. Human–chimpanzee comparison also makes it possible to study homologous recombination between nearby repeat elements as a source of genomic deletions. We found 612 deletions (totalling 2 Mb) in the human genome that appear to have resulted from recombination between two nearby Alu elements present in the common ancestor; there are 914 such events in the chimpanzee genome. (The events are not biased to (A + T)-rich DNA and thus would not explain the preferential loss of Alu elements in such regions discussed above.) Similarly, we found 26 and 48 instances involving adjacent L1 copies and 8 and 22 instances involving retroviral LTRs in human and chimpanzee, respectively. None of the repeat-mediated deletions removed an orthologous exon of a known human gene in chimpanzee.
The genome comparison allows one to estimate the dependency of homologous recombination on divergence and distance. Homologous recombination seems to occur between quite (> 25%) diverged copies (Fig. 8), whereas the number of recombination events (n) varies inversely with the distance (d, in bases) between the copies (as n ≈ 6 × 106 d-1.7; r2 = 0.9).

a, Whereas homologous recombination occurs between quite divergent (Smith–Waterman score <1,000), closely spaced copies, more distant recombination seems to favour a better match between the recombining repeats. b, The frequency of Alu–Alu-mediated recombination falls markedly as a function of distance between the recombining copies. The first three points (magenta) involve recombination between left or right arms of one Alu inserted into another. The high number of occurrences at a distance of 300–400 nucleotides is due to the preference of integration in the A-rich tail; exclusion of this point does not change the parameters of the equation.
Large-scale rearrangements
Finally, we examined the chimpanzee genome sequence for information about large-scale genomic alterations. Cytogenetic studies have shown that human and chimpanzee chromosomes differ by one chromosomal fusion, at least nine pericentric inversions, and in the content of constitutive heterochromatin84. Human chromosome 2 resulted from a fusion of two ancestral chromosomes that remained separate in the chimpanzee lineage (chromosomes 2A and 2B in the revised nomenclature18, formerly chimpanzee chromosomes 12 and 13); the precise fusion point has been mapped and its duplication structure described in detail85,86. In accord with this, alignment of the human and chimpanzee genome sequences shows a break in continuity at this point.
We searched the chimpanzee genome sequence for the precise locations of the 18 breakpoints corresponding to the 9 pericentric inversions (Supplementary Table S22). By mapping paired-end sequences from chimpanzee large insert clones to the human genome, we were able to identify 13 of the breakpoints within the assembly from discordant end alignments. The positions of five breakpoints (on chromosomes 4, 5 and 12) were tested by fluorescence in situ hybridization (FISH) analysis and all were confirmed. Also, the positions of three previously mapped inversion breakpoints (on chromosomes 15 and 18) matched closely those found in the assembly87,88. The paired-end analysis works well in regions of unique sequence, which constitute the bulk of the genome, but is less effective in regions of recent duplication owing to ambiguities in mapping of the paired-end sequences. Beyond the known inversions, we also found suggestive evidence of many additional smaller inversions, as well as older segmental duplications (< 98% identity; Supplementary Fig. S6). However, both smaller inversions and more recent segmental duplications will require further investigations.
Gene evolution
We next sought to use the chimpanzee sequence to study the role of natural selection in the evolution of human protein-coding genes. Genome-wide comparisons can shed light on many central issues, including: the magnitude of positive and negative selection; the variation in selection across different lineages, chromosomes, gene families and individual genes; and the complete loss of genes within a lineage.
We began by identifying a set of 13,454 pairs of human and chimpanzee genes with unambiguous 1:1 orthology for which it was possible to generate high-quality sequence alignments covering virtually the entire coding region (ref. Supplementary Information ‘Gene evolution’ and Table S23). The list contains a large fraction of the entire complement of human genes, although it under-represents gene families that have undergone recent local expansion (such as olfactory receptors and immunoglobulins). To facilitate comparison with the murid lineage, we also compiled a set of 7,043 human, chimpanzee, mouse and rat genes with unambiguous 1:1:1:1 orthology and high-quality sequence alignments (Supplementary Table S24).
Average rates of evolution
To assess the rate of evolution for each gene, we estimated KA, the number of coding base substitutions that result in amino acid change as a fraction of all such possible sites (the non-synonymous substitution rate). Because the background mutation rate varies across the genome, it is crucial to normalize KA for comparisons between genes. A striking illustration of this variation is the fact that the mean KA is 37% higher in the rapidly diverging distal 10 Mb of chromosomes than in the more proximal regions. Classically, the background rate is estimated by KS, the synonymous substitution rate (coding base substitutions that, because of codon redundancy, do not result in amino acid change). Because a typical gene has only a few synonymous changes between humans and chimpanzees, and not infrequently is zero, we exploited the genome sequence to estimate the local intergenic/intronic substitution rate, KI, where appropriate. KA and KS were also estimated for each lineage separately using mouse and rat as outgroups (Fig. 9).

a, Evolutionary tree. The branch lengths are proportional to the absolute rates of amino acid divergence. b, Maximum-likelihood estimates of the rates of evolution in protein-coding genes for humans, chimpanzees, mice and rats. In the text, ωhominid is the KA/KS of the combined human and chimpanzee branches and ωmurid of the combined mouse and rat branches. The slight difference between ωhuman and ωchimpanzee is not statistically significant; masking of some heterozygous bases in the chimpanzee sequence may contribute to the observed difference (see Supplementary Information ‘Gene evolution’).
The KA/KS ratio is a classical measure of the overall evolutionary constraint on a gene, where KA/KS≪1 indicates that a substantial proportion of amino acid changes must have been eliminated by purifying selection. Under the assumption that synonymous substitutions are neutral, KA/KS > 1 implies, but is not a necessary condition for, adaptive or positive selection. The KA/KI ratio has the same interpretation. The ratios will sometimes be denoted below by ω with an appropriate subscript (for example, ωhuman) to indicate the branch of the evolutionary tree under study.
Evolutionary constraint on amino acid sites within the hominid lineage. Overall, human and chimpanzee genes are extremely similar, with the encoded proteins identical in the two species in 29% of cases. The median number of non-synonymous and synonymous substitutions per gene are two and three, respectively. About 5% of the proteins show in-frame indels, but these tend to be small (median = 1 codon) and to occur in regions of repeated sequence. The close similarity of human and chimpanzee genes necessarily limits the ability to make strong inferences about individual genes, but there is abundant data to study important sets of genes.
The KA/KS ratio for the human–chimpanzee lineage (ωhominid) is 0.23. The value is much lower than some recent estimates based on limited sequence data (ranging as high as 0.63 (ref. 7)), but is consistent with an estimate (0.22) from random expressed-sequence-tag (EST) sequencing45. Similarly, KA/KI was also estimated as 0.23.
Under the assumption that synonymous mutations are selectively neutral, the results imply that 77% of amino acid alterations in hominid genes are sufficiently deleterious as to be eliminated by natural selection. Because synonymous mutations are not entirely neutral (see below), the actual proportion of amino acid alterations with deleterious consequences may be higher. Consistent with previous studies8, we find that KA/KS of human polymorphisms with frequencies up to 15% is significantly higher than that of human–chimpanzee differences and more common polymorphisms (Table 3), implying that at least 25% of the deleterious amino acid alterations may often attain readily detectable frequencies and thus contribute significantly to the human genetic load.
Evolutionary constraint on synonymous sites within hominid lineage. We next explored the evolutionary constraints on synonymous sites, specifically fourfold degenerate sites. Because such sites have no effect on the encoded protein, they are often considered to be selectively neutral in mammals.
We re-examined this assumption by comparing the divergence at fourfold degenerate sites with the divergence at nearby intronic sites. Although overall divergence rates are very similar at fourfold degenerate and intronic sites, direct comparison is misleading because the former have a higher frequency of the highly mutable CpG dinucleotides (9% compared with 2%). When CpG and non-CpG sites are considered separately, we find that both CpG sites and non-CpG sites show markedly lower divergence in exonic synonymous sites than in introns (∼50% and ∼30% lower, respectively). This result resolves recent conflicting reports based on limited data sets45,89 by showing that such sites are indeed under constraint.
The constraint does not seem to result from selection on the usage of preferred codons, which has been detected in lower organisms90 such as bacteria91, yeast92 and flies93. In fact, divergence at fourfold degenerate sites increases slightly with codon usage bias (Kendall's τ = 0.097, P < 10-14). Alternatively, the observed constraint at synonymous sites might reflect ‘background selection’—that is, the indirect effect of purifying selection at amino acid sites causing reduced diversity and thereby reduced divergence at closely linked sites42. Given the low rate of recombination in hominid genomes (a 1 kb region experiences only ∼1 crossover per 100,000 generations or 2 million years), such background selection should extend beyond exons to include nearby intronic sites94. However, when the divergence rate is plotted relative to exon–intron boundaries, we find that the rate jumps sharply within a short region of ∼7 bp at the boundary (Fig. 10). This pattern strongly suggests that the action of purifying selection at synonymous sites is direct rather than indirect, suggesting that other signals, for example those involved in splice site selection, may be embedded in the coding sequence and therefore constrain synonymous sites.

Mean divergence around exon boundaries at non-CpG, exonic, fourfold degenerate sites and intronic sites, relative to the closest mRNA splice junction. The divergence rate at exonic, fourfold degenerate sites is significantly lower than at nearby intronic sites (Mann–Whitney U-test; P < 10-27), suggesting that purifying selection limits the rate of synonymous codon substitutions.
Comparison with murids. An accurate estimate of KA/KS makes it possible to study how evolutionary constraint varies across clades. It was predicted more than 30 years ago95 that selection against deleterious mutations would depend on population size, with mutations being strongly selected only if they reduce fitness by s≫1/4N (where N is effective population size). This would predict that genes would be under stronger purifying selection in murids than hominids, owing to their presumed larger population size. Initial analyses (involving fewer than 50 genes96) suggested a strong effect, but the wide variation in estimates of KA/KS in hominids7,8,97 and murids98 has complicated this analysis45.
Using the large collection of 7,043 orthologous quartets, we calculated mean KA/KS values for the various branches of the four-species evolutionary tree (human, chimpanzee, mouse and rat; Fig. 9). The KA/KS ratio for hominids is 0.20. (This is slightly lower than the value of 0.23 obtained with all human–chimpanzee orthologues, probably reflecting slightly greater constraint on the class of proteins with clear orthologues across hominids and murids.)
The KA/KS ratio is markedly lower for murids than for hominids (ωmurid ≈ 0.13 compared with ωhominid ≈ 0.20) (Fig. 9). This implies that there is an ∼35% excess of the amino-acid-changing mutations in the two hominids, relative to the two murids. Excess amino acid divergence may be explained by either increased adaptive evolution or relaxation of evolutionary constraints. As shown in the next section, the latter seems to be the principal explanation.
Relaxed constraints in human evolution. The KA/KS ratio can be used to make inferences about the role of positive selection in human evolution99,100. Because alleles under positive selection spread rapidly through a population, they will be found less frequently as common human polymorphisms than as human–chimpanzee differences8. Positive selection can thus be detected by comparing the KA/KS ratio for common human polymorphisms with the KA/KS ratio for hominid divergence. These ratios have been estimated as ωpolymorphism ≈ 0.20 based on an initial collection of common SNPs in human genes and ωdivergence ≈ 0.34 based on comparison of human and Old World monkey genes8. Thus, the proportion of amino acid changes attributable to positive selection was inferred to be ∼35% (ref. ref. 8). This would imply a huge quantitative role for positive selection in human evolution.
With the availability of extensive data for both human polymorphism and human–chimpanzee divergence, we repeated this analysis (using the same set of genes for both estimates). We find that ωpolymorphism ≈ 0.21–0.23 and ωdivergence ≈ 0.23 are statistically indistinguishable (Table 3). Although some of the amino acid substitutions in human and chimpanzee evolution must surely reflect positive selection, the results indicate that the proportion of changes fixed by positive selection seems to be much lower than the previous estimate8. (Because the previous results involved comparison to Old World monkeys, it is possible that they reflect strong positive selection earlier in primate evolution; however, we suspect that they reflect the fact that relatively few genes were studied and that different genes were used to study polymorphism and divergence.)
Relaxed negative selection pressures thus primarily explain the excess amino acid divergence in hominid genes relative to murids. Moreover, because both ωhuman and ωchimpanzee are similarly elevated this explanation applies equally to both lineages.
We next sought to study variation in the evolutionary rate of genes within the hominid lineage by searching for unusually high or low levels of constraint for genes and sets of genes.
Rapid evolution in individual genes
We searched for individual genes that have accumulated amino acid substitutions faster than expected given the neutral substitution rate; we considered these genes as potentially being under strong positive selection. A total of 585 of the 13,454 human–chimpanzee orthologues (4.4%) have observed KA/KI > 1 (see Supplementary Information ‘Gene evolution’). However, given the low divergence, the KA/KI statistic has large variance. Simulations show that estimates of KA/KI > 1 would be expected to occur simply by chance in at least 263 cases if purifying selection is allowed to act non-uniformly across genes (Supplementary Fig. S7).
Nonetheless, this set of 585 genes may be enriched for genes that are under positive selection. The most extreme outliers include glycophorin C, which mediates one of the Plasmodium falciparum invasion pathways in human erythrocytes101; granulysin, which mediates antimicrobial activity against intracellular pathogens such as Mycobacterium tuberculosis102; as well as genes that have previously been shown to be undergoing adaptive evolution, such as the protamines and semenogelins involved in reproduction103 and the Mas-related gene family involved in nociception104. With similar follow-up studies on candidates from this list, one may be able to draw conclusions about positive selection on other individual genes. In subsequent sections, we examine the rate of divergence for sets of related genes with the aim of detecting subtler signals of accelerated evolution.
Variation in evolutionary rate across physically linked genes
We explored how the rate of evolution varies regionally across the genome. Several studies of mammalian gene evolution have noted that the rate of amino acid substitution shows local clustering, with proteins encoded by nearby genes evolving at correlated rates16,105,106,107.
Variation across chromosomes. On the basis of an analysis of ∼100 genes108, it was recently reported that the normalized rate of protein evolution is greater on the nine chromosomes that underwent major structural rearrangement during human evolution (chromosomes 1, 2, 5, 9, 12, 15, 16, 17 and 18); it was suggested that such rearrangements led to reduced gene flow and accelerated adaptive evolution. A subsequent study of a collection of chimpanzee ESTs gave contradictory results109,110. With our larger data set, we re-examined this issue and found no evidence of accelerated evolution on chromosomes with major rearrangements, even if we considered each rearrangement separately (Supplementary Table S25).
Among all hominid chromosomes, the most extreme outlier is chromosome X with a mean KA/KI of 0.32. The higher mean seems to reflect a skewed distribution at both high and low values, with the median value (0.17) being more in line with other chromosomes (0.15). The excess of low values may reflect greater purifying selection at some genes, owing to the hemizygosity of chromosome X in males. The excess of high values may reflect increased adaptive selection also resulting from hemizygosity, if a considerable proportion of advantageous alleles are recessive111. Interestingly, the higher KA/KI value on the X chromosome versus autosomes is largely restricted to genes expressed in testis83.
Variation in local gene clusters. We next searched for genomic neighbourhoods with an unusually high density of rapidly evolving genes. Specifically, we calculated the median KA/KI for sliding windows of ten orthologues and identified extreme outliers (P < 0.001 compared to random ordering of genes; see Supplementary Information ‘Gene evolution’). A total of 16 such neighbourhoods were found, which greatly exceeds random expectation (Table 4). Repeating the analysis with larger windows (25, 50 and 100 orthologues) did not identify additional rapidly diverging regions.
In nearly all cases, the regions contain local clusters of phylogenetically and functionally related genes. The rapid diversification of gene families, postulated by ref. 112, can thus be readily discerned even at the relatively close distance of human–chimpanzee divergence. Most of the clusters are associated with functional categories such as host defence and chemosensation (see below). Examples include the epidermal differentiation complex encoding proteins that help form the cornified layer of the skin barrier (Supplementary Fig. S8), the WAP-domain cluster encoding secreted protease inhibitors with antibacterial activity, and the Siglec cluster encoding CD33-related genes. Rapid evolution in these clusters does not seem to be unique to either human or chimpanzee113,114.
Variation in evolutionary rate across functionally related genes
We next studied variation in the evolutionary rate of functional categories of genes, based on the Gene Ontology (GO) classification115.
Rapidly and slowly evolving categories within the hominid lineage. We started by searching for sets of functionally related genes with exceptionally high or low constraint in humans and chimpanzees. For each of the 809 categories with at least 20 genes, KA/KS was calculated by concatenating the gene sequences. The category-specific ratios were compared to the average across all orthologues to identify extreme outliers using a metric based on the binomial test (Supplementary Information ‘Gene evolution’ and Supplementary Tables S26–S29). The numbers of observed outliers below a specific threshold (test statistic <0.001) were then compared to the expected distribution of outliers given randomly permuted annotations.
A total of 98 categories showed elevated KA/KS ratios at the specified threshold (Table 5). Only 30 would be expected by chance, indicating that most (but not all) of these categories undergo significantly accelerated evolution relative to the genome-wide average (P < 10-4). The rapidly evolving categories within the hominid lineage are primarily related to immunity and host defence, reproduction, and olfaction, which are the same categories known to be undergoing rapid evolution within the broader mammalian lineage, as well as more distantly related species15,16,116. Hominids thus seem to be typical of mammals in this respect (but see below).
A total of 251 categories showed significantly low KA/KS ratios (compared with ∼32 expected by chance; P < 10-4). These include a wide range of processes including intracellular signalling, metabolism, neurogenesis and synaptic transmission, which are evidently under stronger-than-average purifying selection. More generally, genes expressed in the brain show significantly stronger average constraint than genes expressed in other tissues83.
Differences between hominid and murid lineages. Having found gene categories that show substantial variation in absolute evolutionary rate within hominids, we next examined variation in relative rates between murids and hominids. The KA/KS of each of the GO categories are highly correlated between the hominid and murid orthologue pairs, suggesting that the selective pressures acting on particular functional categories have been largely proportional in recent hominid and recent murid evolution (Fig. 11). However, there are several categories with significantly accelerated non-synonymous divergence on each of the lineages, which might represent functions that have undergone lineage-specific positive selection or a lineage-specific relaxation of constraint (Supplementary Information ‘Gene evolution’ and Supplementary Tables S30–S39).

GO categories with putatively accelerated (test statistic <0.001; see Methods) non-synonymous divergence on the hominid lineages (red) and on the murid lineages (orange) are highlighted. Owing to the hierarchical nature of GO, the categories do not all represent independent data points. A non-redundant list of significant categories is provided in Table 8 and a complete list in Supplementary Table S30.
A total of 59 categories (compared with 11 expected at random, P < 0.0003) show evidence of accelerated non-synonymous divergence in the murid lineage. These are dominated by functions and processes related to host defence, such as immune response and lymphocyte activation. Examples include genes encoding interleukins and various T-cell surface antigens (Cd4, Cd8, Cd80). Combined with the recent observation that genes involved in host defence have undergone gene family expansion in murids16,17, this suggests that the immune system has undergone extensive lineage-specific innovation in murids. Additional categories that also show relative acceleration in murids include chromatin-associated proteins and proteins involved in DNA repair. These categories may have similarly undergone stronger adaptive evolution in murids or, alternatively, they may contain fewer sites for mutations with slightly deleterious effects (with the result that the KA/KS ratios are less affected by the differences in population size96,117).
Another 58 categories (versus 14 expected at random, P < 0.0005) show evidence of accelerated evolution in hominids, with the set dominated by genes encoding proteins involved in transport (for example, ion transport), synaptic transmission, spermatogenesis and perception of sound (Table 6). Notably, some outliers include genes with brain-related functions, compatible with a recent finding118. Potential positive selection on spermatogenesis genes in the hominids was also recently noted119. However, as above, it is possible that these categories could have more sites for slightly deleterious mutations and thus be more affected by population size differences. Sequence information from more species and from individuals within species will be necessary to distinguish between the possible explanations.
Differences between the human and chimpanzee lineage. One of the most interesting questions is perhaps whether certain categories have undergone accelerated evolution in humans relative to chimpanzees, because such genes might underlie unique aspects of human evolution.
As was done for hominids and murids above, we compared non-synonymous divergence for each category to search for relative acceleration in either lineage (Fig. 12). Seven categories show signs of accelerated evolution on the human lineage relative to chimpanzee, but this is only slightly more than the four expected at random (P < 0.22). Intriguingly, the single strongest outlier is ‘transcription factor activity’, with the 348 human genes studied having accumulated 47% more amino acid changes than their chimpanzee orthologues. Genes with accelerated divergence in human include homeotic, forkhead and other transcription factors that have key roles in early development. However, given the small number of changes involved, additional data will be required to confirm this trend. There was no excess of accelerated categories on the chimpanzee lineage.

GO categories with putatively accelerated (test statistic <0.001; see Methods) non-synonymous divergence on the human lineage (red) and on the chimpanzee lineage (orange) are highlighted. The variance of these estimates is larger than that seen in the hominid–murid comparison owing to the small number of lineage-specific substitutions. Owing to the hierarchical nature of the GO ontology, the categories do not all represent independent data points. A complete list of categories is provided in Supplementary Table S30.
We also compared human genes with and without disease associations, including mental retardation, for differences in mutation rate when compared to chimpanzee. Briefly, no significant differences were observed in either the background mutation rate or in the ratio of human-specific changes to chimpanzee-specific amino acid changes (see Supplementary Information ‘Gene evolution’ and Supplementary Tables S40 and S41).
We thus find minimal evidence of acceleration unique to either the human or chimpanzee lineage across broad functional categories. This is not simply due to general lack of power resulting from the small number of changes since the divergence of human and chimpanzee, because one can detect acceleration of categories in either hominid relative to either murid. For example, 29 accelerated categories versus 9 expected at random (P < 0.02) can be detected on the human lineage, and 40 categories versus 11 expected at random (P < 0.007) on the chimpanzee lineage, relative to mouse. But the outliers are largely the same for both human and chimpanzee, indicating that the fraction of amino acid mutations that have contributed to human- and chimpanzee-specific patterns of evolution must be small relative to the fraction that have contributed to a common hominid and, to a large extent, mammalian pattern of evolution.
It was recently reported10 that several functional categories are enriched for genes with evidence of positive selection in the human lineage or the chimpanzee lineage, and that these categories are largely different between the two lineages. These results and ours differ in ways that will require further investigation. With the potential exception of some developmental regulators, the categories that ref. 10 reported as showing the strongest enrichment of positive selection in one lineage (including cell adhesion, ion transport and perception of sound) are among those that we show as having accelerated divergence in both human and chimpanzee. This suggests that positive selection and relaxation of constraints may be correlated, or alternatively, that the results of ref. 10 may be enriched for false positives in categories that have experienced particularly strong relaxation of constraints in the hominids. Data from additional primates, as well as advances in analytical methods, will be necessary to distinguish between these alternatives. At present, strong evidence of positive selection unique to the human lineage is thus limited to a handful of genes120.
Our analysis above largely omitted genes belonging to large gene families, because gene family expansion makes it difficult to define 1:1:1:1 orthologues across hominids and murids. One of the largest such families, the olfactory receptors, is known to be undergoing rapid divergence in primates. Directed study of these genes in the draft assembly has suggested that more than 100 functional human olfactory receptors are likely to be under no evolutionary constraint121. Our analysis also omitted the majority of very recently duplicated genes owing to their lower coverage in the current chimpanzee assembly. However, recent human-specific duplications can be readily identified from the finished human genome sequence, and have previously been shown to be highly enriched for the same categories found to have high absolute rates of evolution in 1:1 orthologues here; that is, olfaction, immunity and reproduction23.
Gene disruptions in human and chimpanzee
Whereas most genes have undergone only subtle substitutions in their amino acid sequence, a few dozen have suffered more marked changes. We found a total of 53 known or predicted human genes that are either deleted entirely (36) or partially (17) in chimpanzee (Supplementary Table S42). We have so far tested and confirmed 15 of these cases by polymerase chain reaction (PCR) or Southern blotting. An additional eight genes have sustained large deletions (> 15 kb) entirely within an intron. Some genes may have been missed in this count owing to limitations of the draft genome sequence. In addition, some genes may have suffered chain termination mutations or altered reading frames in chimpanzee, but accurate identification of these will require higher-quality sequence. The sensitivity of the reciprocal analysis of genes disrupted in human is currently limited by the small number of independently predicted gene models for the chimpanzee. Some of the gene disruptions may be related to interesting biological differences between the species, as discussed below.
Genetic basis for human- and chimpanzee-specific biology
Given the substantial number of neutral mutations, only a small subset of the observed gene differences is likely to be responsible for the key phenotypic changes in morphology, physiology and behavioural complexity between humans and chimpanzees. Determining which differences are in this evolutionarily important subset and inferring their functional consequences will require additional types of evidence, including information from clinical observations and model systems122. We describe some novel examples of genetic changes for which plausible functional or physiological consequences can be suggested.
Apoptosis. Mouse and human are known to differ with respect to an important mediator of apoptosis, caspase-12 (refs 123–125). The protein triggers apoptosis in response to perturbed calcium homeostasis in mice, but humans seem to lack this activity owing to several mutations in the orthologous gene that together affect the protein produced by all known splice forms; the mutations include a premature stop codon and a disruption of the SHG box required for enzymatic activity of caspases. By contrast, the chimpanzee gene encodes an intact open reading frame and SHG box, indicating that the functional loss occurred in the human lineage. Intriguingly, loss-of-function mutations in mice confer increased resistance to amyloid-induced neuronal apoptosis without causing obvious developmental or behavioural defects126. The loss of function in humans may contribute to the human-specific pathology of Alzheimer's disease, which involves amyloid-induced neurotoxicity and deranged calcium homeostasis.
Inflammatory response. Human and chimpanzee show a notable difference with respect to important mediators of immune and inflammatory responses. Three genes (IL1F7, IL1F8 and ICEBERG) that act in a common pathway involving the caspase-1 gene all appear to be deleted in chimpanzee. ICEBERG is thought to repress caspase-1-mediated generation of pro-inflammatory IL1 cytokines, and its absence in chimpanzee may point to species-specific modulation of the interferon-γ- and lipopolysaccharide-induced inflammatory response127.
Parasite resistance. Similarly, we found that two members of the primate-specific APOL gene cluster (APOL1 and APOL4) have been deleted from the chimpanzee genome. The APOL1 protein is associated with the high-density lipoprotein fraction in serum and has recently been proposed to be the lytic factor responsible for resistance to certain subspecies of Trypanosoma brucei, the parasite that causes human sleeping sickness and the veterinary disease nagana128. The loss of the APOL1 gene in chimpanzees could thus explain the observation that human, gorilla and baboon possess the trypanosome lytic factor, whereas the chimpanzee does not129.
Sialic acid biology related proteins. Sialic acids are cell-surface sugars that mediate many biological functions130. Of 54 genes involved in sialic acid biology, 47 were suitable for analysis. We confirmed and extended findings on several that have undergone human-specific changes, including disruptions, deletions and domain-specific functional changes113,131,132. Human- and chimpanzee-specific changes were also found in otherwise evolutionarily conserved sialyl motifs in four sialyl transferases (ST6GAL1, ST6GALNAC3, ST6GALNAC4 and ST8SIA2), suggesting changes in donor and/or acceptor binding130. Lineage-specific changes were found in a complement factor H (HF1) sialic acid binding domain associated with human disease133. Human SIGLEC11 has undergone gene conversion with a nearby pseudogene, correlating with acquisition of human-specific brain expression and altered binding properties134.
Human disease alleles
We next sought to identify putative functional differences between the species by searching for instances in which a human disease-causing allele appears to be the wild-type allele in the chimpanzee. Starting from 12,164 catalogued disease variants in 1,384 human genes, we identified 16 cases in which the altered sequence in a disease allele matched the chimpanzee sequence, and had plausible support in the literature (Table 7; see also Supplementary Table S43). Upon re-sequencing in seven chimpanzees, 15 cases were confirmed homozygous in all individuals, whereas one (PON1 I102V) appears to be a shared polymorphism (Supplementary Table S44).
Six cases represent de novo human mutations associated with simple mendelian disorders. Similar cases have also been found in comparisons of more distantly related mammals135, as well as between insects136, and have been interpreted as a consequence of a relatively high rate of compensatory mutations. If compensatory mutations are more likely to be fixed by positive selection than by neutral drift136, then the variants identified here might point towards adaptive differences between humans and chimpanzees. For example, the ancestral Thr 29 allele of cationic trypsinogen (PRSS1) causes autosomal dominant pancreatitis in humans137, suggesting that the human-specific Asn 29 allele may represent a digestion-related molecular adaptation138.
The remaining ten cases represent common human polymorphisms that have been reported to be associated with complex traits, including coronary artery disease and diabetes mellitus. In all of these cases we confirmed that the disease-associated allele in humans is indeed the ancestral allele by showing that it is carried not only by chimpanzee but also by outgroups such as the macaque. These ancestral alleles may thus have become human-specific risk factors due to changes in human physiology or environment, and the polymorphisms may represent ongoing adaptations. For example, PPARG Pro 12 is the wild-type allele in chimpanzee but has been clearly associated with increased risk of type 2 diabetes in human139. It is tempting to speculate that this allele may represent an ancestral ‘thrifty’ genotype140.
The current results must be interpreted with caution, because few complex disease associations have been firmly established. The fact that the human disease allele is the wild-type allele in chimpanzee may actually indicate that some of the putative associations are spurious and not causal. However, this approach can be expected to become increasingly fruitful as the quality and completeness of the disease mutation databases improve.
Human population genetics
The chimpanzee has a special role in informing studies of human population genetics, a field that is undergoing rapid expansion and acquiring new relevance to human medical genetics141. The chimpanzee sequence allows recognition of those human alleles that represent the ancestral state and the derived state. It also allows estimates of local mutation rates, which serve as an important baseline in searching for signs of natural selection.
Ancestral and derived alleles
Of ∼7.2 million SNPs mapped to the human genome in the current public database, we could assign the alleles as ancestral or derived in 80% of the cases according to which allele agrees with the chimpanzee genome sequence142 (see Supplementary Information ‘Human population genetics’). For the remaining cases, no assignment could be made because of the following: the orthologous chimpanzee base differed from both human alleles (1.2%); was polymorphic in the chimpanzee sequences obtained (0.4%); or could not be reliably identified with the current draft sequence of the chimpanzee (18.8%), with many of these occurring in repeated or segmentally duplicated sequence. The first two cases arise presumably because a second mutation occurred in the chimpanzee lineage. It should be possible to resolve most of these cases by examining a close outgroup such as gorilla or orang-utan.
Mutations in the chimpanzee may also lead to the erroneous assignment of human alleles as derived alleles. This error rate can be estimated as the probability of a second mutation resulting in the chimpanzee sequence matching the derived allele (see Supplementary Information ‘Human population genetics’). The estimated error rate for typical SNPs is 0.5%, owing to the low nucleotide substitution rate. The exceptions are those SNPs for which the human alleles are CpG and TpG and the chimpanzee sequence is TpG. For these, a non-negligible fraction may have arisen by two independent deamination events within an ancestral CpG dinucleotide, which are well-known mutational hotspots51 (also see above). Human SNPs in a CpG context for which the orthologous chimpanzee sequence is TpG account for 12% of the total, and have an estimated error rate of 9.8%. Across all SNPs, the average error rate, ɛ, is thus estimated to be ∼1.6%.
We compared the distribution of allele frequencies for ancestral and derived alleles using a database of allele frequencies for ∼120,000 SNPs (see Supplementary Information ‘Human population genetics’). As expected, ancestral alleles tend to have much higher frequencies than derived alleles (Supplementary Fig. S9). Nonetheless, a significant proportion of derived alleles have high frequencies: 9.1% of derived alleles have frequency ≥80%.
An elegant result in population genetics states that, for a randomly interbreeding population of constant size, the probability that an allele is ancestral is equal to its frequency143. We explored the extent to which this simple theoretical expectation fits the human population. We tabulated the proportion pa(x) of ancestral alleles for various frequencies of x and compared this with the prediction pa(x) = x (Fig. 13).

The solid line shows the regression (b = 0.83). The dotted line shows the theoretical relationship pa(x) = x. Note that because each variant yields a derived and an ancestral allele, the data are necessarily symmetrical about 0.5.
The data lie near the predicted line, but the observed slope (0.83) is substantially less than 1. One explanation for this deviation is that some ancestral alleles are incorrectly assigned (an error rate of ɛ would artificially decrease the slope by a factor of 1–2ɛ). However, with ɛ estimated to be only 1.6%, errors can only explain a small part of the deviation. The most likely explanation is the presence of bottlenecks during human history, which tend to flatten the distribution of allele frequencies. Theoretical calculations indicate that a recent bottleneck would decrease the slope by a factor of (1 - b), where b is the inbreeding coefficient induced by the bottleneck (see Supplementary Information ‘Human population genetics’ and Supplementary Fig. S10). This suggests that measurements of the slope in different human groups may shed light on population-specific bottlenecks. Consistent with this, preliminary analyses of allele frequencies in several regions for SNPs obtained by systematic uniform sampling indicate that the slope is significantly lower than 1 in European and Asian samples and close to 1 in an African sample (see Supplementary Information ‘Human population genetics’ and Supplementary Fig. S11).
Signatures of strong selective sweeps in recent human history
The pattern of human genetic variation holds substantial information about selection events that have shaped our species. Strong positive selection creates the distinctive signature of a ‘selective sweep’, whereby a rare allele rapidly rises to fixation and carries the haplotype on which it occurs to high frequency (the ‘hitchhiking’ effect). The surrounding region should show two distinctive signatures: a significant reduction of overall diversity, and an excess of derived alleles with high frequency in the population owing to hitchhiking of derived alleles on the selected haplotype (see Supplementary Information ‘Human population genetics’). The pattern might be detectable for up to 250,000 years after a selective sweep has ended144. Notably, the chimpanzee genome provides crucial baseline information required for accurate assessment of both signatures.
The size of the interval affected by a selective sweep is expected to scale roughly with s, the selective advantage due to the mutation. Simulations can be used to study the distribution of the interval size (see Supplementary Information ‘Human population genetics’). With s = 1%, the interval over which heterozygosity falls by 50% has a modal size of 600 kb and a probability of greater than 10% of exceeding 1 Mb.
We undertook an initial scan for large regions (> 1 Mb) with the two signatures suggestive of strong selective sweeps in recent human history. We began by identifying regions in which the observed human diversity rate was much lower than the expectation based on the observed divergence rate with chimpanzee. The human diversity rate was measured as the number of occurrences from a database of 1.92 million SNPs identified by shotgun sequencing in a panel of African–American individuals (see Supplementary Information ‘Genome sequencing and assembly’). The comparison with the chimpanzee eliminates regions in which low diversity simply reflects a low mutation rate in the region. Regions were identified based on a simple statistical procedure (see Supplementary Information ‘Human population genetics’). Six genomic regions stand out as clear outliers that show significantly reduced diversity relative to divergence (Table 8; see also Supplementary Fig. S12).
We next tested whether these six regions show a high proportion of SNPs with high-frequency derived alleles (defined here as alleles with frequency ≥80%). Within each region, we focused on the 1-Mb interval with the greatest discrepancy between diversity and divergence and compared it to 1-Mb regions throughout the genome. For the database of 120,000 SNPs with allele frequencies discussed above, the typical 1-Mb region in the human genome contains ∼40 SNPs, and the proportion ph of SNPs with high-frequency derived alleles is ∼9.1%. All six regions identified by our scan for reduced diversity have a higher than average fraction of high-frequency derived alleles; all six fall within the top 10% genome-wide and three fall within the top 1%. Although this is not definitive evidence for any particular region, the joint probability of all six regions randomly scoring in the top 10% is 10-6. The results indicate that the six regions are candidates for strong selective sweeps during the past 250,000 years144. The regions differ notably with respect to gene content, ranging from one containing 57 annotated genes (chromosome 22) to another with no annotated genes whatsoever (chromosome 4). We have no evidence to implicate any individual functional element as a target of recent selection at this point, but the regions contain a number of interesting candidates for follow-up studies. Intriguingly, the chromosome 4 gene desert, which flanks a proto-cadherin gene and is conserved across vertebrates15, has been implicated in two independent studies as being associated with obesity145,146.
In addition to the six regions, one further genomic region deserves mention: an interval of 7.6 Mb on chromosome 7q (see Supplementary Information ‘Human population genetics’). The interval contains several regions with high scores in the diversity-divergence analysis (including the seventh highest score overall) as well as in the proportion of high-frequency derived alleles. The region contains the FOXP2 and CFTR genes. The former has been the subject of much interest as a possible target for selection during human evolution147 and the latter as a target of selection in European populations148.
Convincing proof of past selection will require careful analysis of the precise pattern of genetic variation in the region and the identification of a likely target of selection. Nonetheless, our findings suggest that the approach outlined here may help to unlock some of the secrets of recent human evolution through a combination of within-species and cross-species comparison.
Discussion
Our knowledge of the human genome is greatly advanced by the availability of a second hominid genome. Some questions can be directly answered by comparing the human and chimpanzee sequences, including estimates of regional mutation rates and average selective constraints on gene classes. Other questions can be addressed in conjunction with other large data sets, such as issues in human population genetics for which the chimpanzee genome provides crucial controls. For still other questions, the chimpanzee genome simply provides a starting point for further investigation.
The hardest such question is: what makes us human? The challenge lies in the fact that most evolutionary change is due to neutral drift. Adaptive changes comprise only a small minority of the total genetic variation between two species. As a result, the extent of phenotypic variation between organisms is not strictly related to the degree of sequence variation. For example, gross phenotypic variation between human and chimpanzee is much greater than between the mouse species Mus musculus and Mus spretus, although the sequence difference in the two cases is similar. On the other hand, dogs show considerable phenotypic variation despite having little overall sequence variation (∼0.15%). Genomic comparison markedly narrows the search for the functionally important differences between species, but specific biological insights will be needed to sift the still-large list of candidates to separate adaptive changes from neutral background.
Our comparative analysis suggests that the patterns of molecular evolution in the hominids are typical of a broader class of mammals in many ways, but distinctive in certain respects. As with the murids, the most rapidly evolving gene families are those involved in reproduction and host defence. In contrast to the murids, however, hominids appear to experience substantially weaker negative selection; this probably reflects their smaller population size. Consequently, hominids accumulate deleterious mutations that would be eliminated by purifying selection in murids. This may be both an advantage and a disadvantage. Although decreased purifying selection may tend to erode overall fitness, it may also allow hominids to ‘explore’ larger regions of the fitness landscape and thereby achieve evolutionary adaptations that can only be reached by passing through intermediate states of inferior fitness149,150.
Although the analyses presented here focus on protein-coding sequences, the chimpanzee genome sequence also allows systematic analysis of the recent evolution of gene regulatory elements for the first time. Initial analysis of both gene expression patterns and promoter regions suggest that their overall patterns of evolution closely mirror that of protein-coding regions. In an accompanying paper83, we show that the rates of change in gene expression among different tissues in human and chimpanzee correlate with the nucleotide divergence in the putative proximal promoters and even more interestingly with the average level of constraint on proteins in the same tissues. Another study151 has similarly used the chimpanzee sequence described here to show that gene promoter regions are also evolving under markedly less constraint in hominids than in murids.
The draft chimpanzee sequence here is sufficient for initial analyses, but it is still imperfect and incomplete. Definitive studies of gene and genome evolution—including pseudogene formation, gene family expansion and segmental duplication—will require high-quality finished sequence. In this regard, we note that efforts are already underway to construct a BAC-based physical map and to increase the shotgun sequence coverage to approximately sixfold redundancy. The added coverage alone will not affect the analysis greatly, but plans are in place to produce finished sequence for difficult to sequence and important segments of the genome.
Our close biological relatedness to chimpanzees not only allows unique insights into human biology, it also creates ethical obligations. Although the genome sequence was acquired without harm to chimpanzees, the availability of the sequence may increase pressure to use chimpanzees in experimentation. We strongly oppose reducing the protection of chimpanzees and instead advocate the policy positions suggested by an accompanying paper152. Furthermore, the existence of chimpanzees and other great apes in their native habitats is increasingly threatened by human civilization. More effective policies are urgently needed to protect them in the wild. We hope that elaborating how few differences separate our species will broaden recognition of our duty to these extraordinary primates that stand as our siblings in the family of life.
Methods
Sequencing and assembly
Approximately 22.5 million sequence reads were derived from both ends of inserts (paired end reads) from 4-, 10-, 40- and 180-kb clones, all prepared from primary blood lymphocyte DNA. Genomic resources available from the source animal include a lymphoid cell line (S006006) and genomic DNA (NS06006) at Coriell Cell Repositories (http://locus.umdnj.edu/ccr/), as well as a BAC library (CHORI-251)153 (see also Supplementary Information ‘Genome sequencing and assembly’).
Genome alignment
BLASTZ154 was used to align non-repetitive chimpanzee regions against repeat-masked human sequence. BLAT155 was subsequently used to align the more repetitive regions. The combined alignments were chained156 and only best reciprocal alignments were retained for further analysis.
Insertions and deletions
Small insertion/deletion (indel) events (< 15 kb) were parsed directly from the BLASTZ genome alignment by counting the number and size of alignment gaps between bases within the same contig. Sites of large-scale indels (> 15 kb) were detected from discordant placements of paired sequence reads against the human assembly. Size thresholds were obtained from both human fosmid alignments on human sequence (40 ± 2.58 kb) and chimpanzee plasmid alignments against human chromosome 21 (4.5 ± 1.84 kb). Indels were inferred by two or more pairs surpassing these thresholds by more than two standard deviations and the absence of sequence data within the discordancy.
Gene annotation
A total of 19,277 human RefSeq transcripts157, representing 16,045 distinct genes, were indirectly aligned to the chimpanzee sequence via the genome alignment. After removing low-quality sequences and likely alignment artefacts, an initial catalogue containing 13,454 distinct 1:1 human–chimpanzee orthologues was created for the analyses described here. A subset of 7,043 of these genes with unambiguous mouse and rat orthologues were realigned using Clustal W158 for the lineage-specific analyses. Updated gene catalogues can be obtained from http://www.ensembl.org.
Rates of divergence
Nucleotide divergence rates were estimated using baseml with the REV model. Non-CpG rates were estimated from all sites that did not overlap a CG dinucleotide in either human or chimpanzee. KA and KS were estimated jointly for each orthologue using codeml with the F3x4 codon frequency model and no additional constraints, except for the comparison of divergent and polymorphic substitutions where KA/KS for both was estimated as (ΔA/NA)/(ΔS/NS), with NS/NA, the ratio of synonymous to non-synonymous sites, estimated as 0.36 from the orthologue alignments. Unless otherwise specified, KA/KS for a set of genes was calculated by summing the number of substitutions and the number of sites to obtain KA and KS for the concatenated set before taking the ratio. Hominid and murid pairwise rates were estimated independently from codons aligned across all four species. Human and chimpanzee lineage-specific KA and KS were estimated on an unrooted tree with both mouse and rat included. Lineage-specific rates were also estimated by parsimony, with essentially identical results (see Supplementary Information). KI was estimated from all interspersed repeats within 250 kb of the mid-point of each gene.
Accelerated evolution in GO categories
The binomial probability of observing X or more non-synonymous substitutions, given a total of X + Y substitutions and the expected proportion x from all orthologues, was calculated by summing substitutions across the orthologues in each GO category. For the absolute rate test, Y = the number of synonymous substitutions in orthologues in the same category. For the relative rate tests, Y = the number of non-synonymous substitutions on the opposite lineage. Note that this binomial probability is simply a metric designed to identify potentially accelerated categories, it is not a P-value that can be used to reject directly the null hypothesis of no acceleration in that particular category. For each test, the observed number of categories with a binomial probability less than 0.001 was compared to the expected distribution of such outliers by repeating the procedure 10,000 times on randomly permuted GO annotations. The significance of the number of observed outliers n was estimated as the proportion of random trials yielding n or more outliers.
Detection of selective sweeps
The observed number of human SNPs, ui, human bases, mi, human–chimpanzee substitutions, vi, and chimpanzee bases, ni, within each set of non-overlapping 1-Mb windows along the human genome were used to generate two random numbers, xi (adjusted human diversity) and yi (adjusted human–chimpanzee divergence), from the two beta-distributions:


where a = 1, b = 1,000, c = 1 and d = 100. These numbers were then fit to a linear regression:

A P-value for each window was calculated for each window based on (xi, yi) and the regression line. This was repeated 100 times and the average of the P-values taken as the P-value for diversity given divergence in each window. Overlapping windows with P < 0.1 containing at least one window of P < 0.05 were coalesced and scored as the sum of their -log(p) scores.
The Chimpanzee Sequencing and Analysis Consortium
Tarjei S. Mikkelsen1,2, LaDeana W. Hillier3, Evan E. Eichler4, Michael C. Zody1, David B. Jaffe1, Shiaw-Pyng Yang3, Wolfgang Enard5, Ines Hellmann5, Kerstin Lindblad-Toh1, Tasha K. Altheide6, Nicoletta Archidiacono7, Peer Bork8,9, Jonathan Butler1, Jean L. Chang1, Ze Cheng4, Asif T. Chinwalla3, Pieter deJong10, Kimberley D. Delehaunty3, Catrina C. Fronick3, Lucinda L. Fulton3, Yoav Gilad11, Gustavo Glusman12, Sante Gnerre1, Tina A. Graves3, Toshiyuki Hayakawa6, Karen E. Hayden13, Xiaoqiu Huang14, Hongkai Ji15, W. James Kent16, Mary-Claire King4, Edward J. Kulbokas, III1, Ming K. Lee4, Ge Liu13, Carlos Lopez-Otin17, Kateryna D. Makova18, Orna Man19, Elaine R. Mardis3, Evan Mauceli1, Tracie L. Miner3, William E. Nash3, Joanne O. Nelson3, Svante Pääbo5, Nick J. Patterson1, Craig S. Pohl3, Katherine S. Pollard16, Kay Prüfer5, Xose S. Puente17, David Reich1,20, Mariano Rocchi7, Kate Rosenbloom16, Maryellen Ruvolo21, Daniel J. Richter1, Stephen F. Schaffner1, Arian F. A. Smit12, Scott M. Smith3, Mikita Suyama8, James Taylor18, David Torrents8, Eray Tuzun4, Ajit Varki6, Gloria Velasco17, Mario Ventura7, John W. Wallis3, Michael C. Wendl3, Richard K. Wilson3, Eric S. Lander1,22,23,24, Robert H. Waterston4
Affiliations for participants: 1Broad Institute of MIT and Harvard, 320 Charles Street, Cambridge, Massachusetts 02141, USA. 2Division of Health Sciences and Technology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. 3Genome Sequencing Center, Washington University School of Medicine, Campus Box 8501, 4444 Forest Park Avenue, St Louis, Missouri 63108, USA. 4Genome Sciences, University of Washington School of Medicine, 1705 NE Pacific Street, Seattle, Washington 98195, USA. 5Max Planck Institute of Evolutionary Anthropology, Deutscher Platz 6, D-04103 Leipzig, Germany. 6University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093, USA. 7Department of Genetics and Microbiology, University of Bari, 70126 Bari, Italy. 8EMBL, Meyerhofstrasse 1, Heidelberg D-69117, Germany. 9Max Delbrück Center for Molecular Medicine (MDC), Bobert-Rössle-Strasse 10, D-13125 Berlin, Germany. 10Children's Hospital Oakland Research Institute, 747 52nd Street, Oakland, California 94609, USA. 11Department of Genetics, Yale University School of Medicine, 333 Cedar Street, New Haven, Connecticut 06520, USA. 12Institute for Systems Biology, 1441 North 34th Street, Seattle, Washington 98103, USA. 13Department of Genetics, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, Ohio 44106, USA. 14Department of Computer Science, Iowa State University, 226 Atanasoff Hall, Ames, Iowa 50011, USA. 15Department of Statistics, Harvard University, 1 Oxford Street, Cambridge, Massachusetts 02138, USA. 16University of California, Santa Cruz, Center for Biomolecular Science and Engineering, 1156 High Street, Santa Cruz, California 95064, USA. 17Departamento de Bioquimica y Biologia Molecular, Instituto Universitario de Oncologia del Principado de Asturias, Universidad de Oviedo, C/Fernando Bongera s/n, 33006 Oviedo, Spain. 18The Pennsylvania State University, Center for Comparative Genomics and Bioinformatics and Department of Biology, University Park, Pennsylvania 16802, USA. 19Department of Structural Biology, Weizmann Institute of Science, Rehovot 76100, Israel. 20Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, USA. 21Departments of Anthropology and of Organismic and Evolutionary Biology, Harvard University, 11 Divinity Avenue, Cambridge, Massachusetts 02138, USA. 22Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, USA. 23Whitehead Institute for Biomedical Research, Cambridge, Massachusetts 02142, USA. 24Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA.
References
Darwin, C. The Descent of Man, and Selection in Relation to Sex (D Appleton and Company, New York, 1871)
Huxley, T. H. Evidence as to Man's Place in Nature (Williams and Norgate, London, 1863)
Goodman, M. The genomic record of humankind's evolutionary roots. Am. J. Hum. Genet. 64, 31–39 (1999)
Goodall, J. Tool-using and aimed throwing in a community of free-living chimpanzees. Nature 201, 1264–1266 (1964)
Whiten, A. et al. Cultures in chimpanzees. Nature 399, 682–685 (1999)
Olson, M. V. & Varki, A. Sequencing the chimpanzee genome: insights into human evolution and disease. Nature Rev. Genet. 4, 20–28 (2003)
Eyre-Walker, A. & Keightley, P. D. High genomic deleterious mutation rates in hominids. Nature 397, 344–347 (1999)
Fay, J. C., Wyckoff, G. J. & Wu, C. I. Positive and negative selection on the human genome. Genetics 158, 1227–1234 (2001)
King, M. C. & Wilson, A. C. Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (1975)
Clark, A. G. et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 302, 1960–1963 (2003)
Hellmann, I. et al. Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res. 13, 831–837 (2003)
Ebersberger, I., Metzler, D., Schwarz, C. & Paabo, S. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70, 1490–1497 (2002)
Watanabe, H. et al. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature 429, 382–388 (2004)
Jaillon, O. et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957 (2004)
Hillier, L. W. et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004)
Mouse Genome Sequencing Consortium, Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002)
Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521 (2004)
McConkey, E. H. Orthologous numbering of great ape and human chromosomes is essential for comparative genomics. Cytogenet. Genome Res. 105, 157–158 (2004)
Sanger, F., Coulson, A. R., Hong, G. F., Hill, D. F. & Petersen, G. B. Nucleotide sequence of bacteriophage lambda DNA. J. Mol. Biol. 162, 729–773 (1982)
Myers, G. Whole-genome DNA sequencing. Comput. Sci. Eng. 1, 33–43 (1999)
Huang, X., Wang, J., Aluru, S., Yang, S. P. & Hillier, L. PCAP: a whole-genome assembly program. Genome Res. 13, 2164–2170 (2003)
Jaffe, D. B. et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13, 91–96 (2003)
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004)
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–920 (2001)
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998)
She, X. et al. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature 431, 927–930 (2004)
Cheng, Z. et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature doi:10.1038/nature04000 (this issue)
Fischer, A., Wiebe, V., Paabo, S. & Przeworski, M. Evidence for a complex demographic history of chimpanzees. Mol. Biol. Evol. 21, 799–808 (2004)
Yu, N. et al. Low nucleotide diversity in chimpanzees and bonobos. Genetics 164, 1511–1518 (2003)
Kaessmann, H., Wiebe, V., Weiss, G. & Paabo, S. Great ape DNA sequences reveal a reduced diversity and an expansion in humans. Nature Genet. 27, 155–156 (2001)
Kitano, T., Schwarz, C., Nickel, B. & Paabo, S. Gene diversity patterns at 10 X-chromosomal loci in humans and chimpanzees. Mol. Biol. Evol. 20, 1281–1289 (2003)
The International SNP Map Working Group. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933 (2001)
Chen, F. C. & Li, W. H. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68, 444–456 (2001)
Fujiyama, A. et al. Construction and analysis of a human-chimpanzee comparative clone map. Science 295, 131–134 (2002)
Hardison, R. C. et al. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13, 13–26 (2003)
Webster, M. T., Smith, N. G., Lercher, M. J. & Ellegren, H. Gene expression, synteny, and local similarity in human noncoding mutation rates. Mol. Biol. Evol. 21, 1820–1830 (2004)
Rosenberg, H. F. & Feldmann, M. W. The Relationship Between Coalescence Times and Population Divergence Times (Oxford Univ. Press, Oxford, 2002)
Vignaud, P. et al. Geology and palaeontology of the Upper Miocene Toros-Menalla hominid locality, Chad. Nature 418, 152–155 (2002)
Wall, J. D. Estimating ancestral population sizes and divergence times. Genetics 163, 395–404 (2003)
Reich, D. E. et al. Human genome sequence variation and the influence of gene history, mutation and recombination. Nature Genet. 32, 135–142 (2002)
Maynard Smith, J. M. & Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23–35 (1974)
Hudson, R. R. & Kaplan, N. L. Deleterious background selection with recombination. Genetics 141, 1605–1617 (1995)
Charlesworth, B. The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet. Res. 63, 213–227 (1994)
Birky, C. W. Jr & Walsh, J. B. Effects of linkage on rates of molecular evolution. Proc. Natl Acad. Sci. USA 85, 6414–6418 (1988)
Hellmann, I., Ebersberger, I., Ptak, S. E., Paabo, S. & Przeworski, M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 72, 1527–1535 (2003)
Lercher, M. J. & Hurst, L. D. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18, 337–340 (2002)
Hellmann, I. et al. Why do human diversity levels vary at a megabase scale? Genome Res. 15, 1222–1231 (2005)
Li, W. H., Yi, S. & Makova, K. Male-driven evolution. Curr. Opin. Genet. Dev. 12, 650–656 (2002)
Bohossian, H. B., Skaletsky, H. & Page, D. C. Unexpectedly similar rates of nucleotide substitution found in male and female hominids. Nature 406, 622–625 (2000)
Makova, K. D. & Li, W. H. Strong male-driven evolution of DNA sequences in humans and apes. Nature 416, 624–626 (2002)
Hwang, D. G. & Green, P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl Acad. Sci. USA 101, 13994–14001 (2004)
Taylor, J., Tyekucheva, S., Zody, M., Ciaromonte, F. & Makova, K. D. Strong and weak male mutation bias at different sites in the primate genomes: Insights from the human-chimpanzee comparison. Mol. Biol. Evol. (submitted)
Bulmer, M., Wolfe, K. H. & Sharp, P. M. Synonymous nucleotide substitution rates in mammalian genes: implications for the molecular clock and the relationship of mammalian orders. Proc. Natl Acad. Sci. USA 88, 5974–5978 (1991)
Ehrlich, M., Zhang, X. Y. & Inamdar, N. M. Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat. Res. 238, 277–286 (1990)
Craig, J. M. & Bickmore, W. A. Chromosome bands—flavours to savour. Bioessays 15, 349–354 (1993)
Holmquist, G. P. Chromosome bands, their chromatin flavors, and their functional features. Am. J. Hum. Genet. 51, 17–37 (1992)
Ellegren, H., Smith, N. G. & Webster, M. T. Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev. 13, 562–568 (2003)
Cooper, G. M., Brudno, M., Green, E. D., Batzoglou, S. & Sidow, A. Quantitative estimates of sequence divergence for comparative analyses of mammalian genomes. Genome Res. 13, 813–820 (2003)
Cooper, G. M. et al. Characterization of evolutionary rates and constraints in three mammalian genomes. Genome Res. 14, 539–548 (2004)
Yang, S. et al. Patterns of insertions and their covariation with substitutions in the rat, mouse, and human genomes. Genome Res. 14, 517–527 (2004)
Birdsell, J. A. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol. Biol. Evol. 19, 1181–1197 (2002)
Jensen-Seaman, M. I. et al. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14, 528–538 (2004)
Fortna, A. et al. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2, E207 (2004)
Britten, R. J. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc. Natl Acad. Sci. USA 99, 13633–13635 (2002)
Frazer, K. A. et al. Genomic DNA insertions and deletions occur frequently between humans and nonhuman primates. Genome Res. 13, 341–346 (2003)
Locke, D. P. et al. Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome Res. 13, 347–357 (2003)
Liu, G. et al. Analysis of primate genomic variation reveals a repeat-driven expansion of the human genome. Genome Res. 13, 358–368 (2003)
Yohn, C. T. et al. Lineage-specific expansions of retroviral insertions within the genomes of African great apes but not humans and orangutans. PLoS Biol. 3, 1–11 (2005)
Hedges, D. J. et al. Differential alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res. 14, 1068–1075 (2004)
Smit, A. F. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 9, 657–663 (1999)
Dewannieux, M., Esnault, C. & Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nature Genet. 35, 41–48 (2003)
Mathews, L. M., Chi, S. Y., Greenberg, N., Ovchinnikov, I. & Swergold, G. D. Large differences between LINE-1 amplification rates in the human and chimpanzee lineages. Am. J. Hum. Genet. 72, 739–748 (2003)
Pickeral, O. K., Makalowski, W., Boguski, M. S. & Boeke, J. D. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 10, 411–415 (2000)
Goodier, J. L., Ostertag, E. M. & Kazazian, H. H. Jr Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum. Mol. Genet. 9, 653–657 (2000)
Zhang, Z., Harrison, P. M., Liu, Y. & Gerstein, M. Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 13, 2541–2558 (2003)
Torrents, D., Suyama, M., Zdobnov, E. & Bork, P. A genome-wide survey of human pseudogenes. Genome Res. 13, 2559–2567 (2003)
Esnault, C., Maestre, J. & Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nature Genet. 24, 363–367 (2000)
Zhang, Z., Harrison, P. & Gerstein, M. Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res. 12, 1466–1482 (2002)
Ostertag, E. M., Goodier, J. L., Zhang, Y. & Kazazian, H. H. Jr SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 73, 1444–1451 (2003)
Shen, L. et al. Structure and genetics of the partially duplicated gene RP located immediately upstream of the complement C4A and the C4B genes in the HLA class III region. Molecular cloning, exon-intron structure, composite retroposon, and breakpoint of gene duplication. J. Biol. Chem. 269, 8466–8476 (1994)
Takai, D. & Jones, P. A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl Acad. Sci. USA 99, 3740–3745 (2002)
Enard, W. et al. Intra- and interspecific variation in primate gene expression patterns. Science 296, 340–343 (2002)
Khaitovich, P. et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science(in the press)
Yunis, J. J., Sawyer, J. R. & Dunham, K. The striking resemblance of high-resolution G-banded chromosomes of man and chimpanzee. Science 208, 1145–1148 (1980)
Fan, Y., Linardopoulou, E., Friedman, C., Williams, E. & Trask, B. J. Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and paralogous regions on other human chromosomes. Genome Res. 12, 1651–1662 (2002)
Fan, Y., Newman, T., Linardopoulou, E. & Trask, B. J. Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions. Genome Res. 12, 1663–1672 (2002)
Locke, D. P. et al. Refinement of a chimpanzee pericentric inversion breakpoint to a segmental duplication cluster. Genome Biol. 4, R50 (2003)
Dennehey, B. K., Gutches, D. G., McConkey, E. H. & Krauter, K. S. Inversion, duplication, and changes in gene context are associated with human chromosome 18 evolution. Genomics 83, 493–501 (2004)
Subramanian, S. & Kumar, S. Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome Res. 13, 838–844 (2003)
Duret, L. Detecting genomic features under weak selective pressure: the example of codon usage in animals and plants. Bioinformatics 18 (suppl. 2), S91 (2002)
Sharp, P. M. & Li, W. H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 14, 7737–7749 (1986)
Sharp, P. M., Averof, M., Lloyd, A. T., Matassi, G. & Peden, J. F. DNA sequence evolution: the sounds of silence. Phil. Trans. R. Soc. Lond. B 349, 241–247 (1995)
Moriyama, E. N. & Powell, J. R. Synonymous substitution rates in Drosophila: mitochondrial versus nuclear genes. J. Mol. Evol. 45, 378–391 (1997)
McVean, G. A. et al. The fine-scale structure of recombination rate variation in the human genome. Science 304, 581–584 (2004)
Ohta, T. Slightly deleterious mutant substitutions during evolution. Nature 246, 96–98 (1973)
Ohta, T. Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 40, 56–63 (1995)
Eyre-Walker, A., Keightley, P. D., Smith, N. G. & Gaffney, D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol. Biol. Evol. 19, 2142–2149 (2002)
Makalowski, W. & Boguski, M. S. Synonymous and nonsynonymous substitution distances are correlated in mouse and rat genes. J. Mol. Evol. 47, 119–121 (1998)
McDonald, J. H. & Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (1991)
Sawyer, S. A. & Hartl, D. L. Population genetics of polymorphism and divergence. Genetics 132, 1161–1176 (1992)
Maier, A. G. et al. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nature Med. 9, 87–92 (2003)
Stenger, S. et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282, 121–125 (1998)
Swanson, W. J. & Vacquier, V. D. The rapid evolution of reproductive proteins. Nature Rev. Genet. 3, 137–144 (2002)
Choi, S. S. & Lahn, B. T. Adaptive evolution of MRG, a neuron-specific gene family implicated in nociception. Genome Res. 13, 2252–2259 (2003)
Hardison, R. C. et al. Global predictions and tests of erythroid regulatory regions. Cold Spring Harb. Symp. Quant. Biol. 68, 335–344 (2003)
Lercher, M. J., Chamary, J. V. & Hurst, L. D. Genomic regionality in rates of evolution is not explained by clustering of genes of comparable expression profile. Genome Res. 14, 1002–1013 (2004)
Williams, E. J. & Hurst, L. D. The proteins of linked genes evolve at similar rates. Nature 407, 900–903 (2000)
Navarro, A. & Barton, N. H. Chromosomal speciation and molecular divergence—accelerated evolution in rearranged chromosomes. Science 300, 321–324 (2003)
Zhang, J., Wang, X. & Podlaha, O. Testing the chromosomal speciation hypothesis for humans and chimpanzees. Genome Res. 14, 845–851 (2004)
Lu, J., Li, W. H. & Wu, C. I. Comment on “Chromosomal speciation and molecular divergence-accelerated evolution in rearranged chromosomes”. Science 302, 988 (2003)
Charlesworth, B., Coyne, J. A. & Orr, H. A. Meiotic drive and unisexual hybrid sterility: a comment. Genetics 133, 421–432 (1993)
Ohno, S. Evolution by Gene Duplication (Springer, New York, 1970)
Angata, T., Margulies, E. H., Green, E. D. & Varki, A. Large-scale sequencing of the CD33-related Siglec gene cluster in five mammalian species reveals rapid evolution by multiple mechanisms. Proc. Natl Acad. Sci. USA 101, 13251–13256 (2004)
Teumer, J. & Green, H. Divergent evolution of part of the involucrin gene in the hominoids: unique intragenic duplications in the gorilla and human. Proc. Natl Acad. Sci. USA 86, 1283–1286 (1989)
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet. 25, 25–29 (2000)
Yang, Z. & Bielawski, J. P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503 (2000)
Weinreich, D. M. The rates of molecular evolution in rodent and primate mitochondrial DNA. J. Mol. Evol. 52, 40–50 (2001)
Dorus, S. et al. Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell 119, 1027–1040 (2004)
Neilsen, R. et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3, e170 (2005)
Vallender, E. J. & Lahn, B. T. Positive selection on the human genome. Hum. Mol. Genet. 13 (suppl. 2), R245–R254 (2004)
Gilad, Y., Man, O. & Glusman, G. A comparison of the human and chimpanzee olfactory receptor gene repertoires. Genome Res. 15, 224–230 (2005)
Enard, W. & Paabo, S. Comparative primate genomics. Annu. Rev. Genomics Hum. Genet. 5, 351–378 (2004)
Saleh, M. et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429, 75–79 (2004)
Fischer, H., Koenig, U., Eckhart, L. & Tschachler, E. Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Commun. 293, 722–726 (2002)
Puente, X. S., Sanchez, L. M., Overall, C. M. & Lopez-Otin, C. Human and mouse proteases: a comparative genomic approach. Nature Rev. Genet. 4, 544–558 (2003)
Nakagawa, T. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403, 98–103 (2000)
Humke, E. W., Shriver, S. K., Starovasnik, M. A., Fairbrother, W. J. & Dixit, V. M. ICEBERG: a novel inhibitor of interleukin-1β generation. Cell 103, 99–111 (2000)
Vanhamme, L. et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 422, 83–87 (2003)
Seed, J. R., Sechelski, J. B. & Loomis, M. R. A survey for a trypanocidal factor in primate sera. J. Protozool. 37, 393–400 (1990)
Angata, T. & Varki, A. Chemical diversity in the sialic acids and related α-keto acids: an evolutionary perspective. Chem. Rev. 102, 439–469 (2002)
Varki, A. How to make an ape brain. Nature Genet. 36, 1034–1036 (2004)
Sonnenburg, J. L., Altheide, T. K. & Varki, A. A uniquely human consequence of domain-specific functional adaptation in a sialic acid-binding receptor. Glycobiology 14, 339–346 (2004)
Pangburn, M. K. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology 49, 149–157 (2000)
Hayakawa, T. et al. Human-specific gene in microglia. Science(in the press)
Kondrashov, A. S., Sunyaev, S. & Kondrashov, F. A. Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl Acad. Sci. USA 99, 14878–14883 (2002)
Kulathinal, R. J., Bettencourt, B. R. & Hartl, D. L. Compensated deleterious mutations in insect genomes. Science 306, 1553–1554 (2004)
Pfutzer, R. et al. Novel cationic trypsinogen (PRSS1) N29T and R122C mutations cause autosomal dominant hereditary pancreatitis. Gut 50, 271–272 (2002)
Chen, J. M., Montier, T. & Ferec, C. Molecular pathology and evolutionary and physiological implications of pancreatitis-associated cationic trypsinogen mutations. Hum. Genet. 109, 245–252 (2001)
Altshuler, D. et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genet. 26, 76–80 (2000)
Neel, J. V. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 14, 353–362 (1962)
The International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003)
Hacia, J. G. et al. Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays. Nature Genet. 22, 164–167 (1999)
Watterson, G. A. & Guess, H. A. Is the most frequent allele the oldest? Theor. Popul. Biol. 11, 141–160 (1977)
Przeworski, M. The signature of positive selection at randomly chosen loci. Genetics 160, 1179–1189 (2002)
Stone, S. et al. A major predisposition locus for severe obesity, at 4p15-p14. Am. J. Hum. Genet. 70, 1459–1468 (2002)
Arya, R. et al. Evidence of a novel quantitative-trait locus for obesity on chromosome 4p in Mexican Americans. Am. J. Hum. Genet. 74, 272–282 (2004)
Enard, W. et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418, 869–872 (2002)
Schroeder, S. A., Gaughan, D. M. & Swift, M. Protection against bronchial asthma by CFTR ΔF508 mutation: a heterozygote advantage in cystic fibrosis. Nature Med. 1, 703–705 (1995)
Ohta, T. Evolution by nearly-neutral mutations. Genetica 102–103, 83–90 (1998)
Hayakawa, T., Altheide, T. K. & Varki, A. Genetic basis of human brain evolution: accelerating along the primate speedway. Dev. Cell 8, 2–4 (2005)
Keightley, P. D., Lercher, M. J. & Eyre-Walker, A. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 3, e42 (2005)
Gagneux, P., Moore, J. J. & Varki, A. The ethics of research on great apes. Nature 437, 27–29 (2005)
Osoegawa, K. et al. An improved approach for construction of bacterial artificial chromosome libraries. Genomics 52, 1–8 (1998)
Schwartz, S. et al. Human-mouse alignments with BLASTZ. Genome Res. 13, 103–107 (2003)
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002)
Kent, W. J., Baertsch, R., Hinrichs, A., Miller, W. & Haussler, D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl Acad. Sci. USA 100, 11484–11489 (2003)
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence project: update and current status. Nucleic Acids Res. 31, 34–37 (2003)
Higgins, D. G., Thompson, J. D. & Gibson, T. J. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 266, 383–402 (1996)
Meloni, A. et al. Delineation of the molecular defects in the AIRE gene in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients from Southern Italy. J. Clin. Endocrinol. Metab. 87, 841–846 (2002)
Beales, P. L. et al. Genetic and mutational analyses of a large multiethnic Bardet-Biedl cohort reveal a minor involvement of BBS6 and delineate the critical intervals of other loci. Am. J. Hum. Genet. 68, 606–616 (2001)
Cunningham, J. M. et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am. J. Hum. Genet. 69, 780–790 (2001)
Mukhopadhyay, A. et al. Mutations in MYOC gene of Indian primary open angle glaucoma patients. Mol. Vis. 8, 442–448 (2002)
Tuchman, M., Jaleel, N., Morizono, H., Sheehy, L. & Lynch, M. G. Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum. Mutat. 19, 93–107 (2002)
Clee, S. M. et al. Common genetic variation in ABCA1 is associated with altered lipoprotein levels and a modified risk for coronary artery disease. Circulation 103, 1198–1205 (2001)
Fullerton, S. M. et al. Apolipoprotein E variation at the sequence haplotype level: implications for the origin and maintenance of a major human polymorphism. Am. J. Hum. Genet. 67, 881–900 (2000)
Mentuccia, D. et al. Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the β-3-adrenergic receptor. Diabetes 51, 880–883 (2002)
Pizzuti, A. et al. A polymorphism (K121Q) of the human glycoprotein PC-1 gene coding region is strongly associated with insulin resistance. Diabetes 48, 1881–1884 (1999)
Katoh, T. et al. Human glutathione S-transferase P1 polymorphism and susceptibility to smoking related epithelial cancer; oral, lung, gastric, colorectal and urothelial cancer. Pharmacogenetics 9, 165–169 (1999)
Marchesani, M. et al. New paraoxonase 1 polymorphism I102V and the risk of prostate cancer in Finnish men. J. Natl Cancer Inst. 95, 812–818 (2003)
Humbert, R. et al. The molecular basis of the human serum paraoxonase activity polymorphism. Nature Genet. 3, 73–76 (1993)
Barroso, I. et al. Candidate gene association study in type 2 diabetes indicates a role for genes involved in β-cell function as well as insulin action. PLoS Biol. 1, E20 (2003)
Herrmann, S. M. et al. Uncoupling protein 1 and 3 polymorphisms are associated with waist-to-hip ratio. J. Mol. Med. 81, 327–332 (2003)
Kong, A. et al. A high-resolution recombination map of the human genome. Nature Genet. 31, 241–247 (2002)
Acknowledgements
Generation of the Pan troglodytes sequence at Washington University School of Medicine's Genome Sequencing Center and the Broad Institute of MIT and Harvard was supported by grants from the National Human Genome Research Institute (NHGRI). We would like to thank the entire staff of both of those institutions. For work from other groups, we acknowledge the support of the European Molecular Biology Laboratory, Ministerio de Educacion y Ciencia (Spain), Howard Hughes Medical Institute, NHGRI, National Institutes of Health and National Science Foundation. Resources for exploring the sequence and annotation data are available on browser displays available at UCSC (http://genome.ucsc.edu), Ensembl (http://www.ensembl.org) and the NCBI (http://www.ncbi.nlm.nih.gov). We thank L. Gaffney for graphical help. Author Contributions The last three authors co-directed the work.
Author information
Consortia
Ethics declarations
Competing interests
This Pan troglodytes whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the project accessions ARACHNE, AADA01000000 and PCAP, AACZ01000000. Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
Additional information
Lists of participants and affiliations appear at the end of the paper
Supplementary information
Supplementary Figures
Supplementary Figures S1-S12 and accompanying legends. (PPT 1122 kb)
Supplementary Notes S1
Genome Sequencing and Assembly. Detailed description of chimpanzee DNA and cell-line resources; genome sequencing and assembly methods; assembly quality control; additional discussion. Supplementary Tables S1-S17. (DOC 407 kb)
Supplementary Notes S2
Genome Evolution. Additional methods and discussion related to nucleotide substitution rates, insertions/deletions, transposable elements and large-scale rearrangements. Supplementary Tables S18, S20-S22. (DOC 66 kb)
SupplementaryTable S19
Human- and chimpanzee-specific retrotransposed pseudogenes (XLS 83 kb)
Supplementary Notes S3
Gene Evolution. Additional methods and discussion related to evolutionary rates for protein coding genes, selective constraints, relative rate tests and genetic changes with potential phenotypic effects. Supplementary Tables S25, S27, S29, S31-S42, S44. (DOC 346 kb)
Supplementary Table S23
Rate estimates and additional information for each human-chimpanzee orthologue pair examined. (XLS 3488 kb)
Supplementary Table S24
Rate estimates and additional information for each human-chimpanzee-mouse-rat orthologue quadruplet examined. (XLS 3809 kb)
Supplementary Table S26
Absolute rate acceleration test statistics for each GO category. (XLS 1229 kb)
SupplementaryTable S28
Absolute rate acceleration test statistics (non-parametric) for each GO category. (XLS 806 kb)
Supplementary Table S30
Relative rate acceleration test statistics for each GO category. (XLS 368 kb)
Supplementary Table S43
Putative human disease alleles present in chimpanzee, and status of support in the literature. (XLS 28 kb)
Supplementary Notes S4
Human Population Genetics. Additional methods and discussion related to assignment of ancestral alleles, and detection of selective sweeps in recent human evolution. (DOC 64 kb)
Supplementary Notes S5
Additional References. References cited in the Supplementary Notes. (DOC 30 kb)
Corrigendum
This file contains details regarding an error in Supplementary Table 24 at the time of publication. (DOC 19 kb)
Rights and permissions
About this article
Cite this article
The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). https://doi.org/10.1038/nature04072
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature04072
This article is cited by
-
On the genetic basis of tail-loss evolution in humans and apes
Nature (2024)
-
Emergence and influence of sequence bias in evolutionarily malleable, mammalian tandem arrays
BMC Biology (2023)
-
Evolutionary relevance of single nucleotide variants within the forebrain exclusive human accelerated enhancer regions
BMC Molecular and Cell Biology (2023)
-
Epigenetic regulation of human-specific gene expression in the prefrontal cortex
BMC Biology (2023)
-
Fast-evolving genomic regions underlie human brain development
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
