
Abstract
Strategies for assembling large, complex genomes have evolved to include a combination of whole-genome shotgun sequencing and hierarchal map-assisted sequencing1,2. Whole-genome maps of all types can aid genome assemblies, generally starting with low-resolution cytogenetic maps and ending with the highest resolution of sequence. Fingerprint clone maps are based upon complete restriction enzyme digests of clones representative of the target genome, and ultimately comprise a near-contiguous path of clones across the genome. Such clone-based maps are used to validate sequence assembly order, supply long-range linking information for assembled sequences, anchor sequences to the genetic map and provide templates for closing gaps. Fingerprint maps are also a critical resource for subsequent functional genomic studies, because they provide a redundant and ordered sampling of the genome with clones3. In an accompanying paper4 we describe the draft genome sequence of the chicken, Gallus gallus, the first species sequenced that is both a model organism and a global food source. Here we present a clone-based physical map of the chicken genome at 20-fold coverage, containing 260 contigs of overlapping clones. This map represents approximately 91% of the chicken genome and enables identification of chicken clones aligned to positions in other sequenced genomes.
Similar content being viewed by others


Main
For large genome sequence assemblies so far, contiguous clone-based maps provide the framework for organizing the sequence5,6. With the increasing emphasis on whole-genome shotgun assemblies, integration of mapping data throughout the assembly and finishing process is key to improving efficiency and assuring accuracy equivalent to human genome standards. The basis of map construction for any genome is the reliance on random breakage of genome structure and the subsequent ordering of these genome pieces. Once assembled, physical maps are a key resource in the dissemination of clones by known location for use in all disciplines of biology.
A large number of chicken genomic resources are available, including a collection of large-insert bacterial artificial chromosome (BAC) clone libraries7,8. These libraries were made from the inbred red jungle fowl 256 (RJF, G. gallus) strain used in whole-genome sequencing and from the white leghorn, a domestic breed selected for egg production. Meiotic linkage maps for three mapping populations were used to develop a consensus map containing about 2,000 sequence-tagged site (STS) markers9,10. Markers that were assigned to BACs were a key resource in map construction.
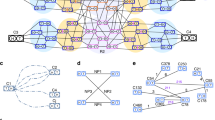
We constructed a physical clone map from 154,560 fingerprints of G. gallus BACs. Automated band recognition software identified restriction digest fragments11. A small fraction (15%) of these fingerprints were lost due to empty lanes, empty insert clones, incomplete restriction enzyme digestion, or failure by the software to recognize lanes. The FPC program processed the remaining 130,486 fingerprints12. Automated overlap evaluation of these fingerprints generated 6,509 contigs using a Sulston score13 threshold of 1 × 10-17, tolerance of 7. The Sulston score approximates the probability of two clones realizing a given number of fingerprint band matches by coincidence. These parameters left 51,954 singleton clones not assigned to a contig. The final automated step refined clone order using the program CORAL14. CORAL has an improved clone-ordering algorithm proven effective when applied to individual FPC contigs.
Subsequent to automated map assembly, manual review is an essential step in building a reliable fingerprint map. Although a required element in high-throughput generation of fingerprints, automated identification of bands produces band-calling errors that cannot be completely eliminated. These errors propagate during clone ordering and result in incorrectly assembled map contigs. Dispersed and tandem sequence repeats can also confound software programs, collapsing different regions into a complex mix of falsely overlapping clones. Finally, there is currently no automated procedure for merging related contigs. Therefore, we visually examined the fingerprint images of digested clones in each contig to limit errors in automated contig construction. Clone order errors were resolved and fingerprints not meeting expectations for band pattern were removed from contigs and returned to the singleton pool. To merge overlapping contigs, we relaxed the Sulston score threshold to 1 × 10-10 and then to 1 × 10-7. Clones comprising the terminal ends of contigs were allowed to join other contigs after manual review. We added individual singleton clones to contigs as needed to increase coverage of sparse regions. If a clone did not provide further band information, it was left in the singleton pool. Of the initial 6,509 contigs, 95% were joined to produce 320 contigs.
Markers are essential for anchoring the fingerprint map to chromosomes, and they also permit validation of contig integrity. A limited number (n = 911) of STS markers have been mapped to fingerprinted RJF clones; 730 of these have chromosome placements15. The white leghorn fingerprint database, however, is a rich source of marker data, with 1,830 markers (1,717 assigned to chromosomes). To increase the number of links to the genetic map, we added the fingerprints of 49,805 white leghorn BAC clones to the physical map. We assigned 29,663 white leghorn clones to contigs, based on the restrictive condition that each of these clones had a fingerprint match to at least five RJF contig clones. Manual inspection confirmed the positions of 2,244 white leghorn clones having assigned markers. The white leghorn clones were used for their marker information only; their fingerprints were not used to join RJF contigs.
We also took advantage of the draft sequence assembly to refine further the fingerprint map. The assembly and fingerprint map are linked by 128,523 BAC-end sequences (BES). After requiring a minimum of six end-sequence links, a total of 189 fingerprint contigs were reliably assigned to the sequence assembly. Some of the contigs left unassigned were linked to assembly contigs in a topologically impossible manner, indicating errors in the assembly or fingerprint map. For example, a pair of contigs cannot be correctly linked by their ends if the middle portions are linked to different contigs. Potentially incorrect regions of the assembly and fingerprint maps were reciprocally examined, revealing 14 misassembled fingerprint contigs that were subsequently split apart. These cases were primarily due to contaminated clones missed during the manual review process.
The assembly also provided preliminary order to 3,557 singleton clones not assigned to fingerprint contigs. Small groups of overlapping singletons often served as bridges that could join contigs. Merges suggested by the assembly were examined and made only when supported by fingerprint data, although a Sulston score of 1 × 10-6 was accepted with the additional sequence evidence. A summary of clone distribution in the final 260 contigs is shown in Table 1.
The genetic map was then used to anchor the fingerprint contigs to chromosomes. We determined the distribution of contigs on each chicken chromosome using a simple plurality of chromosome assignments for the marker–clone pairs in each contig. Markers assigned to white leghorn clones were not considered unless the contig location of the clone had been manually confirmed. A total of 186 contigs were assigned in this way. Linkage of the fingerprint map to the sequence assembly provides positional information for many contigs. All 125 contigs suggested by the assembly to be collinear had consistent chromosome assignments. An additional seven contigs lacking independent marker data were given chromosome assignments based on their linkage to an assigned FPC contig. Finally, markers positioned on assembly contigs by BLAST sequence comparison provided additional support of the chromosome assignments and allowed localization of an additional 33 fingerprint contigs based on their linkage to assembled sequence. A summary of the 226 contigs mapped to chromosomes is found in Table 2.
A minimally overlapping set of clones spanning the map contigs is useful for comparative genomic studies16, as a source of specific cloned sequence, and as a reliable estimator of the physical length of each contig. We used the software Minilda (http://mkweb.bcgsc.ca/minilda/) to select clones with the goals of maximizing the amount of unique content in each clone selection, limiting excessive overlap, avoiding gaps between adjacent selections, and avoiding clones with fingerprint bands unconfirmed by overlapping clones. Our estimated minimum tiling path set consists of 9,210 BAC clones with an average clone overlap of 77 kilobases (kb) (http://mkweb.bcgsc.ca/chicken/images/?list=003). Estimates of the physical size of each contig were made and the totals per chromosome are listed in Table 2. The total amount of sequence represented in fingerprint contigs is 0.97 gigabases (Gb), or 91% of the current sequence assembly4.
Low coverage BAC-based fingerprint maps of the chicken genome were recently published7,8. The maps represent preliminary efforts to generate resources for the community, such as region-specific BAC clones as templates for polymorphic marker development. Ren et al.7 report 2,331 contigs, estimated to cover ∼7.5 × genome equivalents, with 367 markers used to anchor only 11% of the contigs. The estimated 3.6 × genome coverage by Aerts et al.8 was insufficient for accurate comparisons of clone distribution by contig. Both of these reports represented progress towards developing clone resources for functional genomic studies and for improving knowledge of local clone order for selected chromosomes; for example, G. gallus chromosome 10 (GGA10)8. However, neither report created the comprehensive coverage required for sequence assembly validation or provided a sufficient resource to pick tiling paths of reduced genome representation. Our manually curated map of 180,291 clones and 260 contigs represents a major advance upon these early efforts, while at the same time incorporating the identical libraries. In particular, this map integrates an additional, much larger BAC library (CHORI-261), 2,628 STS markers, 128,523 BAC-end sequences, the chicken whole-genome sequence assembly, and much higher clone redundancy. These additional resources allowed us to improve greatly clone contig distribution as demonstrated by a 100-fold increase in large contigs, defined as >200 clones per contig (see Table 1). The physical map is available in standard formats, such as CMAP (http://gmod.wustl.edu/cgi-bin/cmap/viewer) and GBROWSE (http://www.animalsciences.nl/ChickFPC). It can also be downloaded directly from http://genome.wustl.edu/pub/groups/mapping/fpc_files/chicken/.
Combining physical maps with a whole-genome, shotgun-based strategy provides an ideal blueprint for sequence assembly17. Critically, the physical map with corresponding marker information allowed reciprocal error checking of the physical map and the whole-genome shotgun assembly while at the same time providing invaluable long-range linking information. This allowed anchoring, ordering and orienting the sequence along the chicken genome. The clone-based map now also provides a key resource for moving towards improving the chicken genome sequence, filling gaps and sorting out difficult regions of the chicken genome. Future avian molecular genetic research will be greatly aided by this BAC-based map and accompanying minimum tiling path.
Methods
Fingerprinting
Isolation of plasmid DNA from individual BAC clones was performed according to the manufacturer's recommendations (Brinkman Instruments). The HindIII digestion and agarose gel fractionation of BAC clones from TAM31, TAM32 and TAM33 (ref. 7) (http://hbz.tamu.edu/bacindex.html), CHORI-261 (http://bacpac.chori.org) and WAG8 (http://www.zod.wau.nl/abg/) libraries were as described previously18. After fluorescent staining and imaging, gel images were analysed using IMAGE software19 (http://www.sanger.ac.uk/Software/Image), followed by automated band calling with BandLeader11.
BAC end sequencing
End sequences of BAC plasmids were obtained using previously established methods with minor modifications20. Resulting traces were transferred into a database and processed using Autobacend software (S.L., unpublished). All trace files were submitted to the NCBI trace archive (http://www.ncbi.nlm.nih.gov/Traces).
Map construction
Fingerprints were assembled into contigs using FPC12 with a Sulston score threshold of 1 × 10-17 and tolerance of 7. CORAL was used to improve clone order after automated contig builds using FPC. During the manual review of clone order, fingerprint band patterns were compared across a contig. A given band should be found in all correctly positioned clones from a limited region, allowing one to scan a contig from one end to the other, confirming the presence or absence of bands in adjacent clones. An incorrectly positioned clone breaks the overall continuity of banding pattern and was repositioned or removed from the contig. Occasionally, a lane had the fingerprint patterns of two unrelated clones. Clone contamination was confirmed by searching the FPC database for the clone with a similar library name and an exact subset of bands. In this way 576 contaminated clones were removed. A limited number of partially digested clones were also removed from contigs. It is particularly important to order correctly the clones at contig ends. Overlapping contigs are identified by comparing the fingerprints of clones at their ends using successively relaxed Sulston score thresholds. Clones are added from the singleton pool as needed to increase coverage in junction regions when contigs are merged. No attempt was made to add all singleton clones to contigs.
Addition of white leghorn clones
White leghorn clones did not integrate well into contigs made from the larger RJF clones owing to small insert sizes. The white leghorn clones tended to form separate contigs, largely because the Sulston score takes into account total band number as well as the number of bands matching between two clones. Consequently, white leghorn clones were added to the map based only on their fingerprint matches to RJF clones. The criteria for adding white leghorn clones to RJF contigs were Sulston score threshold and number of RJF contig clones matching at that threshold. We examined the number of marker position inconsistencies introduced after adding white leghorn clones to choose appropriate parameters. The heuristically determined requirement of fingerprint matches to a minimum of five RJF contig clones at a Sulston score threshold of 1 × 10-6 limited marker inconsistencies yet added a significant number of white leghorn clones with positional information to contigs. Manual comparisons of white leghorn fingerprints to surrounding clones were limited to the clones that have assigned STS markers.
Fingerprint map linkage to the draft sequence assembly
The threshold of BES links used to declare regions of the fingerprint map and draft sequence collinear is subjective. Our choice of parameters was guided by their impact on the frequency of marker inconsistencies and number of topologically impossible contig combinations. A minimum of six BES links were required before aligning an FPC contig to a sequenced region, provided that 70% of all links were consistent and no topological constraints were violated. With these parameters, the overall frequency of spurious BES links from an FPC contig to non-collinear sequence contigs was 0.10 and the frequency of BES links to sequence contigs that were not aligned to the FPC map was 0.05. Discrepant BES links reveal regions of the FPC map where manual review might be useful. Each FPC contig was scanned using a sliding window of ten BES links, which corresponds to roughly 109 kb (assuming a genome size of 1.06 Gb and a random distribution of BES links). If four or more BES links were to an unaligned sequence contig, that FPC region was flagged for review. Of the 195 flagged regions, 14 contained contaminated clones. The remaining number could be a reflection of the reliability of BES links, errors in fingerprint map not revealed by fingerprints, or errors in the sequence assembly.
References
Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002)
Rat Genome Sequencing Project Consortium. Genome sequence of the brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521 (2004)
Krzywinski, M. et al. A set of BAC clones spanning the human genome. Nucleic Acids Res. 32, 3651–3660 (2004)
International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature doi:10.1038/nature03154 (this issue)
Gregory, S. G. et al. A physical map of the mouse genome. Nature 418, 743–750 (2002)
Krzywinski, M. et al. Integrated and sequence-ordered BAC and YAC-based physical maps for the rat genome. Genome Res. 14, 766–779 (2004)
Ren, C. et al. A BAC-based physical map of the chicken genome. Genome Res. 13, 2754–2758 (2003)
Aerts, J. et al. Integration of chicken genomic resources to enable whole-genome sequencing. Cytogenet. Genome Res. 102, 297–303 (2003)
Groenen, M. A. M. et al. A consensus linkage map of the chicken genome. Genome Res. 10, 137–147 (2000)
Groenen, M. A. M. & Crooijmans, R. P. M. A. in Poultry Genetics, Breeding and Technology (eds Muir, W. M. & Aggrey, S. E.) 497–536 (CAB International, Wallingford, UK, 2003)
Fuhrmann, D. R. et al. Software for automated analysis of DNA fingerprinting gels. Genome Res. 13, 940–953 (2003)
Soderlund, C. H. S., Dunham, A. & French, L. Contigs built with fingerprints, markers, and FPC V4.7. Genome Res. 10, 1772–1787 (2000)
Sulston, J., Mallett, F., Durbin, R. & Horsnell, T. Image analysis of restriction enzyme fingerprint autoradiograms. Comput. Appl. Biosci. 5, 101–106 (1989)
Flibotte, S. et al. Automated ordering of fingerprinted clones. Bioinformatics 20, 1264–1271 (2004)
Romanov, M. N., Price, J. A. & Dodgson, J. B. Integration of animal linkage and BAC contig maps using overgo oligo hybridization. Cytogenet. Genome Res. 102, 277–281 (2003)
Cai, W. et al. Genome-wide detection of chromosomal imbalances in tumors using BAC microarrays. Nature Biotechnol. 20, 393–396 (2002)
Meyers, B. C., Scalabrin, S. & Morgante, M. Mapping and sequencing complex genomes: let's get physical! Nature Rev. Genet. 5, 578–588 (2004)
Marra, M. A. et al. High throughput fingerprint analysis of large-insert clones. Genome Res. 7, 1072–1084 (1997)
Sulston, J. et al. Software for genome mapping by fingerprinting techniques. Comput. Appl. Biosci. 4, 125–132 (1988)
Kelley, J. M. et al. High throughput direct end sequencing of BAC clones. Nucleic Acids Res. 27, 1539–1546 (1999)
Acknowledgements
We thank J. C. Eissenberg, P. Cliften and M. Wendl for critically reading the manuscript. We also thank S. Cornelissen, K. Hemmatian, J. van der Poel, T. Veenendaal and R. Dijkhof for their help fingerprinting the WAG BAC library and, along with J. Price and C. Rondelli, linking BAC clones to markers from the genetic linkage map.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Rights and permissions
About this article
Cite this article
Wallis, J., Aerts, J., Groenen, M. et al. A physical map of the chicken genome. Nature 432, 761–764 (2004). https://doi.org/10.1038/nature03030
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature03030
This article is cited by
-
Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research
Nature Communications (2021)
-
Bursal immunopathology responses of specific-pathogen-free chickens and red jungle fowl infected with very virulent infectious bursal disease virus
Archives of Virology (2018)
-
Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators
Nature Genetics (2017)
-
Reconstruction of gross avian genome structure, organization and evolution suggests that the chicken lineage most closely resembles the dinosaur avian ancestor
BMC Genomics (2014)
-
Condensin I associates with structural and gene regulatory regions in vertebrate chromosomes
Nature Communications (2013)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
