
Abstract
Single-nucleotide polymorphisms (SNPs) in CACNA1C, the α1C subunit of the voltage-gated L-type calcium channel Cav1.2, rank among the most consistent and replicable genetics findings in psychiatry and have been associated with schizophrenia, bipolar disorder and major depression. However, genetic variants of complex diseases often only confer a marginal increase in disease risk, which is additionally influenced by the environment. Here we show that embryonic deletion of Cacna1c in forebrain glutamatergic neurons promotes the manifestation of endophenotypes related to psychiatric disorders including cognitive decline, impaired synaptic plasticity, reduced sociability, hyperactivity and increased anxiety. Additional analyses revealed that depletion of Cacna1c during embryonic development also increases the susceptibility to chronic stress, which suggest that Cav1.2 interacts with the environment to shape disease vulnerability. Remarkably, this was not observed when Cacna1c was deleted in glutamatergic neurons during adulthood, where the later deletion even improved cognitive flexibility, strengthened synaptic plasticity and induced stress resilience. In a parallel gene × environment design in humans, we additionally demonstrate that SNPs in CACNA1C significantly interact with adverse life events to alter the risk to develop symptoms of psychiatric disorders. Overall, our results further validate Cacna1c as a cross-disorder risk gene in mice and humans, and additionally suggest a differential role for Cav1.2 during development and adulthood in shaping cognition, sociability, emotional behavior and stress susceptibility. This may prompt the consideration for pharmacological manipulation of Cav1.2 in neuropsychiatric disorders with developmental and/or stress-related origins.
Similar content being viewed by others


Introduction
Major psychiatric disorders including schizophrenia (SCZ), bipolar disorder (BPD), major depressive disorder (MDD) and autism are moderately to highly heritable, and increasing evidence is suggesting a shared genetic etiology among them.1, 2, 3, 4, 5, 6 This might partially explain why many patients suffering from these illnesses display a substantial amount of overlapping symptoms.7 One of the most consistent and robust genetic findings from genome-wide association studies (GWAS) and meta-analyses of GWAS are associations of single-nucleotide polymorphisms (SNPs) in the α1 subunit (CACNA1C) of the voltage-gated L-type Ca2+ channel (LTCC) Cav1.2 with SCZ and BPD, and to a lesser extent with MDD and autism.1, 8, 9, 10, 11, 12, 13, 14, 15, 16 In support, candidate analysis studies have clearly indicated a shared genetic risk for CACNA1C across these disorders.1, 15, 17 Further evidence comes from clinical studies, which have associated the primary disease-associated CACNA1C risk allele, rs1006737, with variations in human brain function and structure in patients, but also in healthy subjects.18, 19, 20 Hence, the available GWAS and clinical data establish CACNA1C as a possible shared susceptibility factor which influences disease vulnerability for BPD, SCZ and MDD across current diagnostic boundaries. This is of considerable interest, in view of the fact that LTCCs have a pivotal role in modulating neuronal excitability, synaptic plasticity and gene expression.21, 22, 23 However, the causality and mechanisms of how genetic alterations in CACNA1C affect the risk for an entire spectrum of psychiatric disorders remain largely unknown. Importantly, the associated CACNA1C variants by themselves only confer a marginal increase in disease risk. In addition to shared genetic risk factors, the environment represents an important contributor to the risk for many psychiatric disorders. In particular, adverse life events such as severe trauma and/or chronic stress represent strong risk factors not only for MDD but also for other psychiatric disorders including BPD and SCZ.24, 25, 26 However, no study to date has examined whether the established risk gene CACNA1C interacts with established environmental risk factors to shape disease outcome.
Functional LTCCs are hetero-oligomeric complexes consisting of multiple subunits: α1, β, α2, δ and/or γ. The voltage sensor, selectivity filter, ion-conduction pore and binding site for all available calcium channel blockers is encoded by the α1 subunit. The LTCC family consists of four distinct members, Cav1.1–Cav1.4, with mainly Cav1.2 and Cav1.3 shown to have a prominent role in the brain,19, 22, 23 and Cav1.2 accounting for ~85% of the LTCCs.27 Pharmacological agents, such as the LTCC-targeting dihydropyridines, have frequently been applied to assess the function of LTCCs in the central nervous system (CNS). However, this approach is limited by the fact that all LTCC antagonists available to date are not completely selective for either Cav1.2 or Cav1.3, and that most studies investigated acute rather than long-term effects. More sophisticated genetic approaches using transgenic mice have helped to address the selective functional roles of different LTCCs.19, 22, 23 Constitutive deletion of Cacna1c was shown to result in embryonic lethality28 and conditional inactivation of Cav1.2 in forebrain structures was repeatedly associated with impairments in cognitive function.29, 30, 31, 32 More recently, haploinsufficiency and forebrain-specific deletion of Cacna1c were also associated with anxiety-like behavior33, 34 and sleep disturbances,35 two core endophenotypes of MDD and BPD. In addition, Cacna1c was shown to mediate survival of young hippocampal neurons36 and brain-specific deletion of Cacna1c impairs discrete forms of hippocampal-dependent memory and neurogenesis within the dentate gyrus.37 However, the underlying neuronal circuits that modulate the effects of CACNA1C on synaptic plasticity and behavior remain largely unknown.
In this study we aimed to investigate how CACNA1C modulates the risk to psychiatric disorders in dependence of environmental, developmental and circuit specific factors, using both genetic mouse models and human data.
Materials and methods
For a detailed description of the Materials and Methods, please refer to the Supplementary Information.
Animals
Male mice were used for all experiments. Inactivation of Cacna1c from forebrain glutamatergic neurons during development was achieved by breeding Cacna1clox/lox mice29 to Nex-Cre mice,38 to obtain Cav1.2-DevGlu-Ctrl (Cacna1clox/lox) and Cav1.2-DevGlu-CKO (Cacna1clox/lox:Nex-Cre). Homozygous and heterozygous deletion of Cacna1c from forebrain excitatory projection neurons in adulthood was achieved by breeding Cacna1clox/lox mice to transgenic Camk2α-CreERT2 mice39 to obtain Cav1.2-AdGlu-Ctrl (Cacna1clox/lox) and Cav1.2-AdGlu-CKO (Cacna1lox/lox:Camk2α-CreERT2), as well as Cav1.2-AdGlu-Ctrl (Cacna1c+/lox) and Cav1.2-AdGlu-Het (Cacna1+/lox:Camk2α-CreERT2), respectively. Cacna1c inactivation in Cav1.2-AdGlu-CKO and Cav1.2-AdGlu-Het mice was induced via 2 weeks of tamoxifen-containing food administration initiated during postnatal weeks 11–13. Both, control (Ctrl) and conditional knockout (CKO; CKOhet) mice received the identical tamoxifen diet. Behavioral experiments were started following an additional two-week washout period in which Cav1.2-AdGlu-CKO and Cav1.2-AdGlu-Het mice received regular chow. Adult male CNS-specific Cacna1c knockout mice and Ctrl littermates were obtained by initially breeding Cacna1clox/lox CKO with Nestin-Cre mice.40 Subsequently Cacna1clox/lox mice were bred to Cacna1c−/+:Nestin-Cre mice to obtain Cav1.2CNS-Ctrl (Cacna1c+/lox:Nestin-Cre) and Cav1.2CNS-CKO (Cacna1c−/lox:Nestin-Cre). Heterozygous Cav1.2 mice and their Ctrl littermates were obtained from the same breedings; Cav1.2Ctrl (Cacna1c+/lox) and Cav1.2Het (Cacna1c−/lox). All animals were kept under standard laboratory conditions and were maintained on a 12 h light–dark cycle (lights on from 0700 to 1900 h), with food and water provided ad libitum. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the Government of Upper Bavaria, Germany.
Chronic social defeat stress paradigm
The chronic social defeat stress (CSDS) paradigm is commonly applied to induce anxiety- and depression-related endophenotypes in mice, and was performed as previously described.41, 42, 43 See also Supplementary Information.
Behavioral testing and study design
Locomotion and sociability were investigated with the open field (OF) test and the three chamber apparatus respectively, as previously described.41, 44 Anxiety-related behaviour was assessed in the light/dark box test as previously described.45 Spatial memory learning and object recognition were investigated with the water cross maze (WCM)46 and spatial object recognition task. The forced swim test (FST) was used to assess passive vs active stress-coping behavior (behavioral despair) and corticosterone levels in response to an acute stressor as previously described.41 For details, refer to online Supplementary Information.
For the basal behavioral characterization of Cav1.2-DevGlu-CKO (Figure 2), Cav1.2-AdGlu-CKO (Figure 2) and Cav1.2CNS-CKO mice (Supplementary Figure S7), one batch of animals was used for each line with the following order of tests (OF, sociability, dark/light box test, FST and corticosterone assessment). A second and third batch of animals was used for the WCM test and long-term potentiation (LTP) recordings, respectively. In addition, a fourth batch of Cav1.2-AdGlu-CKO mice (Figure 4) was used for the CSDS paradigm with the following test order (sociability, OF, object recognition test, dark/light box, FST and subsequent corticosterone, adrenal gland and thymus weight assessment). The same applies to Cav1.2-AdGlu-Het mice (Supplementary Figure S9). For the CSDS experiment in Cav1.2Het mice (Figure 3), two separate batches of animals were used. For batch 1, the OF and dark/light box test were performed. For batch 2, the following tests were performed in this order: sociability, object recognition test, FST and subsequent corticosterone, adrenal gland and thymus weight assessment.
Electrophysiology
The influence of Cacna1c deficiency on hippocampal LTP was conducted as previously described.47 For details, please refer to online Supplementary Information.
Single and double in situ hybridization
Single and double in situ hybridization (ISH) was performed as previously described.48 The Cacna1c probe was designed to target the loxP flanked exons 14 and 15 (nucleotides 2307–2427 of GenBank accession number NM_009781). In addition, the following riboprobes were used: Gad67: 984–1940 of NM_008077; Gad65: 753–1600 of NM_008078; Vglut1 (Slc17a7): 1716–2332 of NM_010484; Vglut2 (Slc17a6): 2427–3006 of NM_080853.3; and LacZ: 2649–3281 of X65335. Images were analyzed with ImageJ (http://rsweb.nih.gov/ij/). Three to four independent experiments were performed for all ISHs and double ISHs.
Study design (gene × environment interaction in humans)
Participants in this study belonged to a larger cohort (Grady Trauma Project) investigating the role of genetic and environmental factors in predicting outcomes to stressful life events.49, 50, 51 Phenotypes and genotypes were available for a total of 4,023 individuals, predominantly African Americans belonging to a highly traumatized, urban population of low socioeconomic status. All procedures were approved by the institutional review boards of Emory University School of Medicine and Grady Memorial Hospital. Please see Supplementary Methods for further details and statistics.
Statistical analysis
Statistical analyses were performed using the commercially available software SPSS v16.0 (SPSS, Chicago, IL, USA) and GraphPad Prism v5.0 (GraphPad Software, La Jolla, CA, USA). The sample size was chosen such that with a type 1 error of 0.05 and a type 2 error of 0.2 the effect size should be at least 1.2-fold of the pooled s.d. All results are presented as mean±s.e.m. Behavioral phenotypic differences between two genotypes were evaluated with Student’s t-test (two-tailed). If data were not normally distributed, the non-parametric Mann–Whitney test was used. Time-dependent measures were assessed with multi-factorial analysis of variance (ANOVA) with repeated measures. For CSDS experiments, the effects of genotype and condition on all other behavioral and neuroendocrine parameters were assessed by two factorial ANOVA (two-way ANOVA). Whenever significant main or interaction effects were found by the ANOVAs, Bonferroni post hoc tests were carried out to locate simple effects. Statistical significance was defined as P<0.05. All data were tested for outlieres using the Grubb’ test. Homogeneity of variance was tested using Bartlett’s test. Animals were allocated to the experimental groups in a semi-randomized manner and data analysis was performed blinded to the group allocation.
Results
Forebrain Cacna1c is predominately expressed in glutamatergic neurons
Cacna1c is expressed throughout the mouse brain, including key limbic regions relevant for emotion and cognition such as the prefrontal cortex (Ctx), hippocampus (Hip) and amygdala (Figure 1a). Double ISH against Cacna1c and markers of excitatory glutamatergic (Vglut1 and Vglut2), as well as inhibitory GABAergic neurons (Gad65/Gad67), revealed a predominant expression of Cacna1c in glutamatergic Vglut1-positive neurons throughout the Ctx, Hip and basolateral amygdala. The prominent Cacna1c expression in the thalamus was largely restricted to glutamatergic Vglut2-positive neurons, although a number of Cacna1c-containing neurons within the latero-dorsal thalamus also co-expressed Vglut1 (Figures 1b and d and Supplementary Figure S1e,f). Minimal to no co-expression with Vglut1 was detected in the caudate putamen, central nucleus of the amygdala, bed nucleus of the stria terminalis or granule cell layer of the olfactory bulb, confirming the abundance of GABAergic markers in these regions. Accordingly, Cacna1c expression in the olfactory bulb, CPu, bed nucleus of the stria terminalis and central nucleus of the amygdala was mainly restricted to Gad65/67-positive GABAergic neurons. In addition, Cacna1c was also detected in scattered Gad65/67-positive neurons of the Hip and Ctx (Figures 1c and d and Supplementary Figure S1c,d).

Cacna1c is predominantly expressed in forebrain glutamatergic neurons. (a) Cacna1c mRNA expression in the mouse brain (C57BL/6J mouse strain), determined by in situ hybridization (ISH). (b and c) Co-localization of Cacna1c with glutamatergic (Vglut1) and GABAergic (Gad65/67) markers with double ISH (DISH). Black arrowheads indicate cells only expressing Cacna1c (silver grains). Gray arrowheads indicate cells co-expressing Cacna1c and the respective neurotransmitter marker (red staining). (d) Quantifications of Cacna1c co-expression with Vglut1 and Gad65/67 (n=3, 2–3 sections per mouse). Scale bar=ISH, 1 mm; DISH, 50 μm.
Deletion of Cacna1c from forebrain glutamatergic neurons during development and adulthood induces differential effects on anxiety and opposing effects on cognition
In view of the results described above, we set out to genetically dissect the role of Cacna1c in glutamatergic neurons. For this, we crossed floxed Cacna1c mice to Nex-Cre mice,38 in which Cre-mediated recombination is initiated during embryonic development (E11.5) in forebrain glutamatergic neurons. As observed from the ISHs and double ISHs, Cacna1c ablation in forebrain glutamatergic neurons (Cav1.2-DevGlu-CKO) was largely restricted to Vglut1-expressing cells, including the Ctx, CA1/2/3 of the Hip, and lateral and basolateral amygdala (Figures 2a and b, and Supplementary Figures S2). Next, we assessed behavioral endophenotypes known to be altered in animal models of MDD, BPD and SCZ, including locomotion/exploration in the OF test, anxiety in the dark/light box test, immobility in the FST, social behavior in the three-chamber test (sociability test), as well as spatial learning and memory in the WCM task. Similar to humans, deficits in cognition and social behavior are recapitulated by mouse models of numerous psychiatric disorders including SCZ, BPD and MDD.52, 53, 54 Hyperactivity in the OF and FST are often associated with mania in mouse models of BPD,53, 55 whereas enhanced anxiety represents a core endophenotype of MDD and the depressive phase of BPD in rodents.52 However, changes in locomotor activity are also commonly reported in animal models of SCZ and MDD.52, 54 Compared with their respective littermate Ctrls, Cav1.2-DevGlu-CKO mice exhibited increased anxiety in the dark–light box test and decreased preference for the social counterpart in the sociability test (Figures 2c and d). In the FST, Cav1.2-DevGlu-CKO mice exhibited increased active stress-coping behavior (reduced behavioral despair), evident by decreased immobility (Figure 2e). In addition, Cav1.2-DevGlu-CKO mice displayed a pronounced hyperlocomotion in the OF test, which was not observed during the initial 5 min where novelty-induced anxiety is most prominent (Figure 2f). The OF was performed under low light conditions (15 lux) in order to minimize anxiety effects on locomotion, which likely explains the lack of effects on inner zone time (Figure 2f). Hip-dependent spatial learning and memory performance was investigated in the WCM.46 Compared with the regular Morris water-maze test, the simplicity of the WCM leads to short trial durations and therefore reduces the stress load compared to Morris water-maze training. In addition, using accuracy rather than speed as the main readout, allows for hippocampus-dependent strategies to be assessed from the first training day on. Cav1.2-DevGlu-CKO mice displayed accuracy levels that barely surpassed the chance level of 50%, both during learning and relearning (Figure 2g), suggesting drastically impaired cognitive performance. In view of this strong effect on cognition, we additionally assessed hippocampal LTP at the Schaffer collateral-CA1 synapses. One hour after a 100 Hz tetanus stimulation, Cav1.2-DevGlu-CKO showed a significant decrease in LTP compared with Ctrl mice (Figure 2h).

Absence of Cacna1c in forebrain glutamatergic neurons during development, but not adulthood, promotes aversive behavioral and cognitive deficits. (a) Cacna1c expression pattern determined by in situ hybridization (ISH) following developmental deletion of Cacna1c (E11.5) in forebrain glutamatergic neurons (Cav1.2-DevGlu-CKO). (b) Double ISH (DISH) in Cav1.2-DevGlu-CKO mice demonstrates absence of Cacna1c mRNA expression (silver grains) in Vglut1-positive neurons (red staining) in the hippocampal CA3 region (conditional knockout (CKO)=3.6±0.74% double-positive neurons normalized to control (Ctrl); one-way analysis of variance (ANOVA): F1,11=365.4, P<0.0001, n=4, 1–2 sections per mouse). Enlarged images of boxed areas are shown in the bottom panel. (c) Dark/light box test (lit time: t26=2.7, P<0.05; No. lit entries: t26=2.2, P<0.05; n=16 Ctrl, 12 CKO). (d) Sociability test (t26=2.9, P<0.05; n=16 Ctrl, 12 CKO). (e) Forced swim test (FST; t26=3.6, P<0.005; n=16 Ctrl, 12 CKO). (f) Distance travelled an inner zone time in the open field (OF) test (distance segments: repeated measures (RM)-ANOVA:time × genotype: F5,130=5.7, P<0.0001; genotype: F(1,130)=45.9, P<0.001; total distance: t26=6.8, P<0.0001; n=16 Ctrl, 12 CKO). (g) Water cross-maze test (RM-ANOVA: genotype: F1,88=7.5, P<0.05; time × genotype: F4,88=7.3, P<0.0001; n=13 Ctrl, 11 CKO). (h) Schaffer collateral/CA1-long-term potentiation (LTP) in Cav1.2-DevGlu-CKOmice (fEPSP last 10 min: t24=2.36, P<0.05; n=14 slices from 4 Ctrl mice and 12 slices from 4 CKO mice (i) Cacna1c expression pattern determined by ISH following adult-specific deletion (postnatal week 11–13) of Cacna1c in forebrain glutamatergic neurons (Cav1.2-AdGlu-CKO). (j) DISHs in Cav1.2-AdGlu-CKO mice demonstrated the absence of Cacna1c mRNA expression in Vglut1-positive neurons in the hippocampal CA3 region (CKO=4.5±1.6% double-positive neurons normalized to Ctrl; one-way ANOVA: F1,11=365.4, P<0.0001, n=4, 1–2 sections per mouse). Enlarged images of boxed areas are shown in the bottom panel. (k–m) Dark/light box, sociability and FST (t=trend, P=0.09, n=17 Ctrl, 11 CKO) in Cav1.2-AdGlu-CKO mice. (n) OF test (distance segments: RM-ANOVA: genotype, F1,130=9.06, P<0.05; total distance: t26=3.01, P<0.05; n=17 Ctrl, 11 CKO) in Cav1.2-AdGlu-CKO mice. (o) Water cross-maze test (RM-ANOVA: genotype, F1,80=4.36, P<0.05; n=11 Ctrl, 10 CKO) in Cav1.2-AdGlu-CKO mice. (p) Schaffer collateral/CA1-LTP in Cav1.2-AdGlu-CKO mice (fEPSP last 10 min: t27=2.06, P<0.05; n=13 slices from 6 Ctrl mice and 16 slices from 7 CKO mice). Student’s t-test for simple comparisons, *P<0.05. Data are means±s.e.m.
Next we addressed whether the underlying neurobiological changes responsible for the behavioral alterations in Cav1.2-DevGlu-CKO mice occur during development or adulthood. Floxed Cacna1c mice were bred to inducible Camk2α-CreERT2 animals39 with the aim of deleting Cacna1c in forebrain glutamatergic neurons (Cav1.2-AdGlu-CKO) during adulthood (postnatal weeks 11–13). We deliberately chose the Camk2α-CreERT2 line because of the strongly overlapping expression pattern with the Nex-Cre line (Figures 2a and i, and Supplementary Figures S2 and S5). Cav1.2 was inactivated upon tamoxifen administration in forebrain excitatory projection neurons, which predominantly include glutamatergic pyramidal neurons of the Ctx, Hip and basolateral amygdala.39 As expected, the mRNA deletion pattern strongly resembled that of Cav1.2-DevGlu-CKO mice, with loss of Cacna1c expression in Vglut1-positive neurons of the lateral and basolateral amygdala, as well as the entire cerebral Ctx and Hip (Figures 2i and j, and Supplementary Figures S3 and S4). Along these lines, remaining hippocampal protein levels did not significantly differ between Cav1.2-DevGlu-CKO and Cav1.2-AdGlu-CKO(Supplementary Figure S6a,b). In addition, deletion of Cacna1c mRNA expression in Cav1.2-DevGlu-CKO mice was also observed throughout the DG and within a few neurons of the latero-dorsal thalamus and medial parts of the thalamus, central nucleus of the amygdala and the geniculate nucleus (Supplementary Figures S2). In the OF, Cav1.2-AdGlu-CKO mice displayed enhanced locomotion (Figure 2n), although this was not as strongly pronounced as in Cav1.2-DevGlu-CKO mice. However, immobility in the FST and sociability were not significantly altered in Cav1.2-DevGlu-CKO mice (Figures 2l and m). Surprisingly, Cav1.2-AdGlu-CKO mice displayed a marginal increase in time spent in the lit zone and number of entries in the dark/light box test (Figure 2k), and even demonstrated enhanced cognitive flexibility during the relearning trial of the WCM (Figure 2o). Considering that deletion of forebrain Cacna1c was reported to impair long-term memory,32 we re-exposed the animals to the WCM 30 days after relearning, but did not observe any differences between Cav1.2-AdGlu-CKO and Ctrl mice (Supplementary Figure S6g). In view of our results that developmental inactivation of Cacna1c from glutamatergic neurons decreases NMDA receptor (NMDAR)-dependent synaptic plasticity and impairs spatial memory, we additionally assessed hippocampal LTP in Cav1.2-AdGlu-CKO mice. Intriguingly, 1 h after a 100 Hz tetanus stimulation, Cav1.2-AdGlu-CKO showed increased LTP compared with Ctrl mice (Figure 2p). Importantly, we observed that CNS-specific Cacna1c deletion (Cav1.2CNS-CKO mice), which is initiated at E8,56 results in similar behavioral alterations compared with Cav1.2-DevGlu-CKO mice (Supplementary Figure S7). This further supports partially opposing roles for developmental and adult Cacna1c in emotion and cognition, and argues against the possibility that differences between Cav1.2-DevGlu-CKO and Cav1.2-AdGlu-CKO mice are due to the slight discrepancies in recombination patterns between the Nex-Cre and Camk2a-CreERT2. In addition, the observed behavioral changes were not caused by alterations in hypothalamic-pituitary-adrenal axis function in neither of the mouse lines (Supplementary Figures S6c,d).
Cacna1c differentially modulates susceptibility to CSDS during development and adulthood
Chronic stress and/or trauma represent strong risk factors for a number of psychiatric disorders, including MDD, BPD and anxiety disorders such as post-traumatic stress disorder.24, 25, 26, 57 In view of the phenotype in Cav1.2-DevGlu-CKO mice, we wondered whether developmental deletion of Cacna1c would increase the susceptibility to CSDS. We choose to investigate heterozygous Cacna1c animals (Cav1.2Het, Figure 3a), considering that Cav1.2-DevGlu-CKO mice already display strong behavioral deficits under baseline conditions, which might not be further aggravated by CSDS. Importantly, no gross behavioral abnormalities were previously reported for Cav1.2Het mice.33 Cav1.2Ctrl and Cav1.2Hetmice were subjected to 3 weeks of CSDS. In accordance with other studies,41, 42 we were able to detect robust physiological and neuroendocrine changes evoked by CSDS, independent of genotype, demonstrating the efficacy of the paradigm (Supplementary Figures S8a–d). Chronically stressed Cav1.2Het mice displayed drastically reduced locomotion throughout the entire test duration compared to both, non-stressed Cav1.2Het and stressed Cav1.2Ctrl mice (Figure 3b). In addition, only chronically stressed Cav1.2Het mice showed a significant reduction in the number of inner zone entries of the OF, indicating increased anxiety-related behavior even under low-light test conditions (Figure 3c). Moreover, CSDS increased the latency to enter the lit compartment and decreased the time spent in the lit zone and number of lit entries to a much greater extent in Cav1.2Het than in Cav1.2Ctrl mice (Figures 3d–f), further supporting increased stress-induced anxiety in heterozygous Cacna1c mice. Sociability, immobility in the FST and spatial object recognition memory were not differentially affected by CSDS (Figures 3g–i), suggesting a specific impact of the interaction between Cacna1c and chronic stress on anxiety.

Cacna1c heterozygosis induces susceptibility to chronic social defeat stress (CSDS). (a) Cacna1c in situ hybridization (ISH) in control (Ctrl) and Cav1.2Het (Het) mice, in which heterozygosis is present from the first day of development. (b) Locomotion (distance segments: time × genotype × stress, F(2,41)=4.3, P=0.02/total distance: genotype, F1,42=23.6, P<0.0001; stress, F1,42=25.4, P<0.0001; genotype × stress, F1,42=4.9, P=0.033) and (c) number of inner zone entries (stress × genotype, F1,42=5.1, P=0.03; stress, F1,42=18.1, P<0.0001; genotype, F1,42=16.3, P=0.0002) in the open field (OF) test (n=12 Ctrl basal, 15 Ctrl stress, 10 Het basal, 9 Het stress). (d–f) Dark/light box test (Lit zone time: stress × genotype, F1,44=8.40, P=0.006; stress, F1,44=21.2, P<0.0001; genotype, F1,44=6.6, P=0.01/Lit zone entries: stress, F1,44=26.1, P<0.0001; genotype, F1,44=16.3, P<0.0001; n=14 Ctrl basal, 15 Ctrl stress, 10 Het basal, 9 Het stress). (g–i) Sociability test (stress, F1,35=8.0, P=0.008), forced swim test (FST; genotype, F1,35=5.6, P=0.01) and spatial object recognition test (n=9 Ctrl basal, 10 Ctrl stress, 10 Het basal, 10 Het stress). Two-way analysis of variance (ANOVA)+Bonferroni post hoc test and repeated measures (RM)-ANOVA+Bonferroni post hoc test; *significantly different from the Ctrl group of the same condition, #significantly different from the basal condition of the same genotype. Data are means±s.e.m.
In view of the partially opposing effects of Cacna1c on anxiety and cognition during development and adulthood, we wondered whether stress vulnerability would also be differentially affected upon deletion of the calcium channel in adulthood. Physiological and neuroendocrine parameters were similarly altered by CSDS in Ctrl and Cav1.2-AdGlu-CKO mice (Supplementary Figures 8e–h). In contrast to Cav1.2Het mice, locomotion/exploration in the OF was only decreased in stressed Ctrl but not stressed Cav1.2-AdGlu-CKO mice, with a similar trend in the number of inner zone entries (Figures 4a and b). In the dark/light box test, only Ctrl mice responded to CSDS with a significant reduction in time spent in the lit zone and number of entries, and a trend toward delayed latencies (Figure 4c), which implies enhanced resilience to CSDS in Cav1.2-AdGlu-CKO mice. FST behavior, sociability and spatial object were similarly affected by CSDS in Ctrl and Cav1.2-AdGlu-CKO mice (Figures 4d–f). Considering that we observed susceptibility to CSDS in heterozygous Cav1.2 mice, we additionally assessed whether a heterozygous deletion in glutamatergic neurons during adulthood (Cav1.2-AdGlu-Het) would be sufficient to induce stress resilience (Supplementary Figure S9). However, compared with Cav1.2-AdGlu-CKO, Cav1.2-AdGlu-Het mice only exhibited partial resilience to CSDS-induced anxiety in the dark/light box test (Supplementary Figure S9f). This points to a gene-dosage effect and suggests that complete absence of Cacna1c during adulthood, in forebrain glutamatergic neurons, is required to induce the extent of resilience to CSDS observed in Cav1.2-AdGlu-CKOmice.

Deletion of Cacna1c from forebrain glutamatergic neurons during adulthood promotes resilience to chronic social defeat stress (CSDS). (a) Locomotion (distance segments: time × stress, F2,45=10.7, P<0.0001/total distance: genotype, F1,45=19.6, P<0.0001; stress, F1,45=4.3, P=0.04) and (b) number of inner zone entries in the open field (OF). (c) Dark/light box test (Lit zone time: genotype, F1,45=9.3, P=0.004/Lit zone entries: stress, F1,45=5.8, P=0.02; genotype, F1,45=12.3, P<0.001). (d) Sociability test (stress, F1,45=52.52, P<0.0001) and (e) forced swim test (FST; genotype, F1,45=7.5, P=0.009; stress, F1,45=8.3, P=0.006). (f) Spatial object recognition test (stress, F1,42=7.1, P=0.01). Two-way analysis of variance (ANOVA)+Bonferroni post hoc test and repeated measures (RM)-ANOVA+Bonferroni post hoc test; *significantly different from the control (Ctrl) group of the same condition, #significantly different from the basal condition of the same genotype. OF, dark/light box test, sociability and FST (n=13 Ctrl basal, 12 Ctrl stress, 12 conditional knockout (CKO) basal, 12 Het stress). Spatial object recognition test (n=12 Ctrl basal, 10 Ctrl stress, 12 CKO basal, 12 CKO stress). Data are means±s.e.m.
CACNA1C interacts with adult trauma to predict depression symptoms in humans
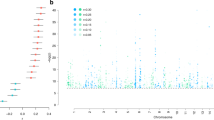
A number of CACNA1C SNPs associated with BPD, SCZ and MDD were also shown to affect measures of anxiety, depression, psychosocial functioning and cognitive aspects in healthy Ctrls,58 which might represent increased susceptibility factors for psychiatric disorders upon exposure to adverse environments. This is especially interesting in the context of MDD, in which disease susceptibility is strongly influenced by previous exposures to chronic and/or severe stress. Considering that most GWAS do not Ctrl for life-time adversity, we wondered whether certain CACNA1C SNPs might primarily influence the risk for MDD in combination with stressful life events. Consequently, we analyzed the effects of all CACNA1C variants and adult trauma on current depression symptoms, determined by the Beck Depression Inventory59 in a large African American cohort of non-psychiatric clinical patients (n=4808) from the Grady trauma project.49, 50, 60 Importantly, symptoms of depression are not only characteristic for MDD and BPD but can also occur as part of affective dysregulation in SCZ24. After linkage disequilibrium-pruning based on r2-values of 0.2, 32 of the 465 tested SNPs in the CACNA1C locus showed nominal significant interactions with adult trauma exposure on Beck Depression Inventory scores. Of these, two remained significant after Bonferroni correction for multiple testing; rs73248708, nominal P=1.38 × 10−5, Bonferroni corrected P=0.0004; rs116625684, nominal P=5.2 × 10−5, Bonferroni corrected P=0.0016 (Figures 5a and b, and Supplementary Table S1 and S2). Both SNPs also showed nominal significant main effects (rs73248708: nominal P=1.88 × 10−4, rs116625684: nominal P=0.02). Interestingly, in both cases, individuals carrying at least one minor allele (CT/TT or AG/AA) and without trauma history displayed significantly higher Beck Depression Inventory scores compared with the major allele group, whereas the opposite was observed for minor allele carriers with the highest degree of lifetime trauma exposure.

CACNA1C interacts with adult trauma to predict depressive symptoms in humans. (a) Single-nucleotide polymorphism (SNP) localization sites in the human CACNA1C locus. Exons are indicated with vertical black bars. (b) rs73249708 (P=0.0004) and rs116625684 (P=0.0016) significantly interacted with adult trauma to predict Beck Depression Inventory scores. Asterisk indicate an interaction P-value below 0.0016, hence surviving Bonferroni correction. The phenotype on adult trauma was categorized into its four quartiles to facilitate interpretation of the plot. Sample size for individual genotypes: rs73248708 (n=4106 GG, 331 AG, 7 AA), rs116625684 (n=4372 AA, 367 AG, 7 GG). Data are means±s.e.m.
Discussion
Our results demonstrate that spatial memory, hippocampal plasticity and anxiety-related behavior are differentially affected by the loss of Cacna1c from excitatory circuits depending on whether it is deleted during embryonic development or adulthood. Increased anxiety-related behavior as well as reduced social and cognitive performance, observed in Cav1.2-DevGlu-CKO mice, are considered core endophenotypes of MDD and/or the depressive phase of BPD,52, 53 whereas increased locomotion and activity in the FST are often associated with mania in animal models of BPD.53, 55 Decreased immobility in the FST was previously reported for heterozygous mice by Dao et al.,33 which is in line with our results and earlier work demonstrating antidepressant-like effects for LTCC blockers.27, 61 However, studies using LTCC blockers have to be interpreted with caution, as these have been reported to induce nonspecific, aversive and stress-like behavioral effects possibly due to excessive blood pressure lowering caused by inhibition of Cav1.2 in the cardiovascular system.62, 63, 64 Importantly, hyperlocomotion, altered social behavior, and learning and memory impairments, are also analogous to negative and cognitive symptoms of SCZ, a disorder believed to have neurodevelopmental origins.24, 54 Additional evidence also suggests that two missense mutations in CACNA1C predispose to autism,65, 66 another neurodevelopmental disorder characterized by impaired social interactions and altered cognition. In accordance with the observed cognitive impairments, Cav1.2-DevGlu-CKO mice also displayed deficits in hippocampal LTP, which is considered the cellular correlate of learning and memory.67 Moreover, we show that Cacna1c strongly interacts with the environment to shape anxiety-related behavior. Heterozygous deletion of Cacna1c, which is present throughout life starting at the earliest point of development (one-cell stage), significantly increased the susceptibility to CSDS and is likely to be mediated by Cacna1c absence in forebrain glutamatergic neurons. Although CACNA1C SNPs have not been consistently associated with anxiety in humans, altered anxiety-related behavior is considered a core endophenotype of MDD and BPD in animal models.52, 53, 55 Different to our observations, a recent study showed that chronic unpredictable stress induces similar behavioral deficits in heterozygous Cav1.2 mice and their wild-type littermates when assessed 1–2 days post stress. Surprisingly, the stress effects persisted in wild-type mice when tested 5–7 days later but not in heterozygous knockout mice.68 These discrepancies might be related to the different chronic stress paradigms.
In contrast to a developmental inactivation, deletion of Cacna1c from forebrain glutamatergic neurons during adulthood blocked the adverse effects of CSDS on anxiety, enhanced LTP and improved hippocampus-dependent cognitive flexibility during the WCM, without affecting long-term memory. Our findings in Cav1.2-DevGlu-CKO mice are supported by previous studies, showing that deletion of Cacna1c from pan-neuronal37 or glutamatergic neurons during development29 impairs synaptic plasticity and/or spatial learning, some of which might be attributed to deficits in adult neurogenesis.37 However, it should be kept in mind that our LTP results cannot be entirely correlated with cognitive performance considering that we stimulated CA3-CA1 Schaffer collaterals, which leads to the potentiation of most CA3-CA1 synapses rather than those that would selectively be recruited during a learning and memory task. Electrophysiological and pharmacological data have indicated that LTP induction is mainly initiated through NMDAR-activity but can also include NMDAR-independent and VGCC-dependent mechanisms.69, 70 Interestingly, the decline of some memory processes is attributed to the shift from NMDAR-dependent to VGCC-intracellular calcium stores-dependent regulation of synaptic plasticity, especially during aging.71, 72, 73 Along these lines, a recent study provides evidence for an association between increased hippocampal Cacna1c expression and age-related cognitive decline.74 Although we did not assess aged animals, it can be speculated that ablation of Cav1.2 in adulthood partially blocks an evolving shift towards VGCC- and/or intracellular calcium store-dependent regulation of synaptic plasticity, hence facilitating NMDAR-dependent LTP. This potentially suggests that Hip-dependent spatial memory formation requires Cav1.2 during prenatal development, but not in adulthood where activation of Cav1.2 channels is even detrimental for LTP. Notably, calcium channel antagonists were shown to ameliorate age-related deficits in hippocampus-dependent memory, possibly mediated through facilitation of NMDAR-LTP or inhibition of long-term depression.71, 73, 75, 76, 77, 78 On the other hand, specific inactivation of Cacna1c in the anterior cingulate between postnatal weeks 8–10, impaired observational fear learning without altering novel object recognition memory and classical fear conditioning,30 highlighting the requirement for Cav1.2 during adulthood for specific cognitive functions. The fact that we observed no significant changes in spatial object memory performance under basal or stress conditions further implies that adult-specific Cacna1c deletion in excitatory circuits does not impact all cognitive domains.
As observed in Cav1.2-DevGlu-CKO mice, locomotion was also enhanced in Cav1.2-AdGlu-CKO animals; however, social and active stress-coping behavior were only mildly affected, suggesting that these endophenotypes are partly regulated by forebrain-Cav1.2 during development and adulthood. In contrast, an earlier study demonstrated that forebrain-specific elimination of Cacna1c, using a non-inducible Camk2α-Cre, also results in increased anxiety-like behavior.34 The discrepancy could potentially be attributed to different Cacna1c deletion time windows (p1879 vs p91-97 in our study).
Although the Nex-Cre and Camk2α-CreERT2 drivers primarily target glutamatergic forebrain neurons, they do slightly differ in their recombination patters (for example,the Camk2α-driven CreERT2 is also expressed in parts of the thalamus, DG and central nucleus of the amygdala). However, the fact that developmental, CNS-specific Cacna1c knockout mice showed similar behavioral deficits as Cav1.2-DevGlu-CKO mice, further supports opposing roles for Cav1.2 during development and adulthood. Nevertheless, we cannot rule out an important contribution of Cav1.2-expressing, GABAergic neurons in many of the disease-associated phenotypes. In fact, Cacna1c was reported to modulate processes related to drug addiction via GABAergic medium spiny neurons in the nucleus accumbens,80, 81 and promote susceptibility to social stress when deleted from this specific brain region.82 It is important to point out that we only assessed a subset of behavioral tests relevant to MDD, BPD and SCZ in our knockout mice. Future studies evaluating anhedonia, psychostimulant-induced locomotion, sleep architecture and deficits in sensorimotor gating (prepulse inhibition deficits), will further refine the contribution of developmental vs adult Cav1.2 in psychiatric disorders.
Given our observations in mice, we further investigated if Cacna1c x stress interactions could also be found in human samples and checked for interaction effects of CACNA1C variants and adult trauma to predict current depressive symptoms
As expected, the risk to develop depressive symptoms was increased with exposure to adult trauma in our study and this effect was moderated by loci within CACNA1C tagged by rs73248708 and rs116625684. Both tag SNPs were not in linkage disequilibrium with the main haplotypes associated with SCZ with genome-wide significance, but represent independent loci. The intron 1 SNP rs116625684 shows a weak, nominal association with SCZ in the Psychiatric Genomics Consortium data set (P=0.021), with an odds ratio of 0.94 (http://www.med.unc.edu/pgc), suggesting a protective effect of the minor allele, which is also indicated by our results for the highest trauma group. The second SNP, rs73248708 is located in intron 3, which harbors the majority of CACNA1C SNPs associated with SCZ at genome-wide significance and also those linked to other disorders including BPD and MDD.19 In fact, SNPs in this region were shown to lie in predicted enhancers capable of physically interacting with the CACNA1C transcriptional start site, and altering gene expression.83 Although rs73248708 is not in linkage disequilibrium with the top GWAS SNPs, including rs1006737, it may independently moderate the function of this enhancer region. In fact, we could show that different sets of genetic variants may be involved in baseline transcriptional regulation vs. regulation following environmental impact, such as trauma exposure.84 Replication in independent cohorts will be necessary to further validate these loci in gene × environment interactions. Importantly, it remains to be investigated whether the here identified SNPs have implications on protein levels or are of other functional relevance. So far, both decreased and increased Cav1.2 levels have been associated with the common risk allele variant rs1006737,85, 86, 87 which might imply different regulatory functions across different brain regions and/or cell types. Whether the here identified SNPs, as well as previously associated CACAN1C risk alleles, can differentially affect gene expression in the brain upon perturbations such as severe stress and/or trauma will have to be assessed in the future. Considering the heterogeneity of MDD, our data raises the possibility that SNPs in CACNA1C might predict the risk for depression mainly in individuals with a history of trauma or severe stress. This could partially explain the nominal associations of CACAN1C with MDD in former genetic studies, which usually do not Ctrl for environmental factors. In addition, given our animal findings it would be interesting to examine for age × trauma interaction effects in the human sample. However, in the present cohort we did not have any information with regard to when exactly adult trauma took place and therefore cannot evaluate age effects.
There are some limitations of the interpretability of our findings, which should not be neglected. First, the Grady Trauma Project includes mostly African Americans. We cannot rule out that effects of CACNA1C variants are different in this specific population as compared with the European population where the effect of CACNA1C variants with regard to MDD were described first. Second, the GRADY sample itself is a highly traumatized cohort. It is possible that the described effects are only present in samples which show a high trauma burden and cannot be found in less traumatized cohorts. However, findings in this cohort can help to transit the results from mice to human data.
Although circuit- or cell type-specific absence of CACNA1C is not likely to occur in humans, epigenetic changes caused by environmental alterations may very well induce expression changes in specific tissues, cell types and/or entire brain circuits. In the clinical context this could imply that, individuals with genetic variants promoting reduced CACNA1C expression may either be at higher or lower risk to develop psychopathologies, depending on whether they experienced severe stress/trauma during development or adulthood. The pronounced effect of a prenatal Cacna1c depletion on emotional behavior and cognitive performance might account for its association with disorders such as SCZ and autism, which are increasingly believed to result from synaptic dysfunction during development. The fact that CACNA1C is also able to interact with the environment during adulthood, by modulating stress susceptibility, could explain its linkage to stress-related disorder such as MDD and BPD. At the same time, Cav1.2 and Cav1.3 are expressed in midbrain dopaminergic neurons and several studies suggest a neuroprotective effects of LTCC blockers in Parkinson’s disease,22, 88 which further emphasizes the possibility that inhibition of Cav1.2 may be of therapeutic value in brain disorders that manifest at later stages in life. Concluding, we propose that the association of CACNA1C with multiple psychiatric disorders is related to its broad expression within key limbic regions and neuronal circuits relevant to emotion, motivation and cognition, and that alterations in CACNA1C gene expression during development and adulthood can result in diverging behavioral outcomes and differentially impact stress susceptibility.
Accession codes
References
Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 2009; 15: 1–7.
Cardno AG, Owen MJ . Genetic relationships between schizophrenia, bipolar disorder, and schizoaffective disorder. Schizophr Bull 2014; 40: 504–515.
Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–94.
Craddock N, O’Donovan MC, Owen MJ . Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr Bull 2006; 32: 9–16.
Crespi B, Stead P, Elliot M . Comparative genomics of autism and schizophrenia. Proc Natl Acad Sci USA 2010; 107: 1736–41.
Sullivan PF, Magnusson C, Reichenberg A, Boman M, Dalman C, Davidson M et al. Family history of schizophrenia and bipolar disorder as risk factors for autism. Arch Gen Psychitary 2012; 69: 1099–1103.
Adam D . On the spectrum. Nature 2013; 496: 416–418.
Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43: 977–83.
Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K et al. Whole-genome association study of bipolar disorder. Mol Psychiatry 2008; 13: 558–569.
Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011; 43: 969–976.
Liu Y, Blackwood DH, Caesar S, de Geus EJC, Farmer A, Ferreira MAR et al. Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Mol Psychiatry 2011; 16: 2–4.
Ferreira MaR, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 2008; 40: 1056–1058.
Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E et al. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry 2009; 14: 252–60.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Smoller JW . Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381: 1371–1379.
Nyegaard M, Demontis D, Foldager L, Hedemand A, Flint TJ, Sørensen KM et al. CACNA1C (rs1006737) is associated with schizophrenia. Mol Psychiatry 2010; 15: 119–121.
Lotan A, Fenckova M, Bralten J, Alttoa A, Dixson L, Williams RW et al. Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Front Neurosci 2014; 8: 331.
Berger SM, Bartsch D . The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 2014; 357: 463–76.
Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol 2012; 99: 1–14.
Ou X, Crane DE, MacIntosh BJ, Young LT, Arnold P, Ameis S et al. CACNA1C rs1006737 genotype and bipolar disorder: focus on intermediate phenotypes and cardiovascular comorbidity. Neurosci Biobehav Rev 2015; 55: 198–210.
Catterall WA . Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 2011; 3: a003947.
Ortner NJ, Striessnig J . L-type calcium channels as drug targets in CNS disorders. Channels 2016; 10: 7–13.
Zamponi GW . Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov 2016; 15: 19–34.
van Os J, Kenis G, Rutten BPF . The environment and schizophrenia. Nature 2010; 468: 203–12.
Kendler K, Karkowski-Shuman L . Stressful life events and genetic liability to major depression: genetic control of exposure to the environment? Psychol Med 1997; 27: 539–547.
Dohrenwend BP, Egri G . Recent stressful life events and episodes of schizophrenia. Schizophr Bull 1981; 7: 12–23.
Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U et al. Expression and 1, 4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 2009; 75: 407–414.
Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem 2000; 275: 39193–39199.
Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 2005; 25: 9883–9892.
Jeon D, Kim S, Chetana M, Jo D, Ruley HE, Lin SY et al. Observational fear learning involves affective pain system and Cav1.2 Ca2+ channels in ACC. Nat Neurosci 2010; 13: 482–488.
Langwieser N, Christel CJ, Kleppisch T, Hofmann F, Wotjak CT, Moosmang S . Homeostatic switch in hebbian plasticity and fear learning after sustained loss of Cav1.2 calcium channels. J Neurosci 2010; 30: 8367–75.
White JA, McKinney BC, John MC, Powers PA, Kamp TJ, Murphy GG . Conditional forebrain deletion of the L-type calcium channel Ca V 1.2 disrupts remote spatial memories in mice. Learn Mem 2008; 15: 1–5.
Dao DT, Mahon PB, Cai X, Kovacsics CE, Blackwell RA, Arad M et al. Mood disorder susceptibility gene CACNA1C modifies mood-related behaviors in mice and interacts with sex to influence behavior in mice and diagnosis in humans. Biol Psychiatry 2010; 68: 801–810.
Lee AS, Ra S, Rajadhyaksha AM, Britt JK, De Jesus-Cortes H, Gonzales KL et al. Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Mol Psychiatry 2012; 17: 1054–5.
Kumar D, Dedic N, Flachskamm C, Voule S, Deussing JM, Kimura M . Cacna1c (Ca(v)1.2) modulates electroencephalographic rhythm and rapid eye movement sleep recovery. Sleep 2015; 38: 1371–1380.
Lee AS, De Jesus-Cortes H, Kabir ZD, Knobbe W, Orr M, Burgdorf C et al. The neuropsychiatric disease-associated gene cacna1c mediates survival of young hippocampal neurons. eNeuro 2016; 3, ENEURO.0006-16.2016.
Temme SJ, Bell RZ, Fisher GL, Murphy GG . Deletion of the mouse homolog of CACNA1C disrupts discrete forms of hippocampal- dependent memory and neurogenesis within the dentate gyrus. eNeuro 2016; 3: e0118–16–14.
Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH . Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 2006; 621: 611–621.
Erdmann G, Schütz G, Berger S . Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci 2007; 8: 63.
Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature 1999; 23: 99–103.
Hartmann J, Wagner KV, Dedic N, Marinescu D, Scharf SH, Wang XD et al. Fkbp52 heterozygosity alters behavioral, endocrine and neurogenetic parameters under basal and chronic stress conditions in mice. Psychoneuroendocrinology 2012; 37: 2009–2021.
Wagner KV, Wang XD, Liebl C, Scharf SH, Müller MB, Schmidt MV . Pituitary glucocorticoid receptor deletion reduces vulnerability to chronic stress. Psychoneuroendocrinology 2011; 36: 579–587.
Hartmann J, Dedic N, Pohlmann ML, Hausl A, Karst H, Engelhardt C et al. Forebrain glutamatergic, but not GABAergic, neurons mediate anxiogenic effects of the glucocorticoid receptor. Mol Psychiatry 2016; 22: 466–475.
Vogl AM, Brockmann MM, Giusti SA, Maccarrone G, Vercelli CA, Bauder CA et al. Neddylation inhibition impairs spine development, destabilizes synapses and deteriorates cognition. Nat Neurosci 2015; 18: 239–251.
Dedic N, Touma C, Romanowski CP, Schieven M, Kühne C, Ableitner M et al. Assessing behavioural effects of chronic HPA axis activation using conditional CRH-overexpressing mice. Cell Mol Neurobiol 2012; 32: 815–28.
Kleinknecht KR, Bedenk BT, Kaltwasser SF, Grünecker B, Yen Y-C, Czisch M et al. Hippocampus-dependent place learning enables spatial flexibility in C57BL6/N mice. Front Behav Neurosci 2012; 6: 87.
Kratzer S, Mattusch C, Metzger MW, Dedic N, Noll-Hussong M, Kafitz KW et al. Activation of CRH receptor type 1 expressed on glutamatergic neurons increases excitability of CA1 pyramidal neurons by the modulation of voltage-gated ion channels. Front Cell Neurosci 2013; 7: 91.
Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM et al. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 2011; 333: 1903–1907.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 2013; 16: 33–41.
Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 2008; 299: 1291–305.
Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA 2013; 110: 8302–7.
Nestler EJ, Hyman SE . Animal models of neuropsychiatric disorders. Nat Neurosci 2010; 13: 1161–9.
Young JW, Henry BL, Geyer MA . Predictive animal models of mania: hits, misses and future directions. Br J Pharmacol 2011; 164: 1263–84.
Pratt J, Winchester C, Dawson N, Morris B . Advancing schizophrenia drug discovery: optimizing rodent models to bridge the translational gap. Nat Rev Drug Discov 2012; 11: 560–79.
Roybal K, Theobold D, Graham A, DiNieri Ja, Russo SJ, Krishnan V et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA 2007; 104: 6406–11.
Dubois NC, Hofmann D, Kaloulis K, Bishop JM, Trumpp A . Nestin-Cre transgenic mouse line Nes-Cre1 mediates highly efficient Cre / loxP mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis 2006; 360: 355–360.
Caspi A, Moffitt TE . Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci 2006; 7: 583–90.
Erk S, Meyer-Lindenberg A, Schnell K, Opitz von Boberfeld C, Esslinger C, Kirsch P et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch Gen Psychiatry 2010; 67: 803–11.
Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J . An inventory for measuring depression. Arch Gen Psychiatry 1961; 4: 561–571.
Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, Gapen M et al. Trauma exposure and stress-related disorders in inner city primary care patients. Gen Hosp Psychiatry 2009; 31: 505–514.
Mogilnicka E, Czyrak A, Maj J . Dihydropyridine calcium channel antagonists reduce immobility in the mouse behavioral despair test; antidepressants facilitate nifedipine action. Eur J Pharmacol 1987; 138: 413–416.
McKinney BC, Sze W, White JA, Murphy GG . L-type voltage-gated calcium channels in conditioned fear: a genetic and pharmacological analysis. Learn Mem 2008; 15: 326–334.
Waltereit R, Mannhardt S, Nescholta S, Maser-Gluth C, Bartsch D . Selective and protracted effect of nifedipine on fear memory extinction correlates with induced stress response. Learn Mem 2008; 15: 348–356.
Busquet P, Hetzenauer A, Sinnegger-brauns MJ, Striessnig J, Singewald N . Role of L-type Ca2+ channel isoforms in the extinction of conditioned fear. Learn Mem 2008; 15: 378–386.
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119: 19–31.
Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW et al. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci USA 2011; 108: 15432–15437.
Kandel ER . The molecular biology of memory storage: a dialogue between gene and synapses. Science 2001; 294: 1030–1038.
Bavley CC, Fischer DK, Rizzo BK, Rajadhyaksha AM . Ca v 1.2 channels mediate persistent chronic stress-induced behavioral deficits that are associated with prefrontal cortex activation of the p25/Cdk5-glucocorticoid receptor pathway. Neurobiol Stress 2017; 7: 27–37.
Grover LM, Teyler TJ . Two components of long-term potentiation induced by different patterns of afferent activation. Nature 1990; 347: 477–479.
Morgan SL, Teyler TJ . VDCCs and NMDARs underlie two forms of LTP in CA1 hippocampus in vivo. J Neurophysiol 1999; 82: 736–740.
Norris CM, Halpain S, Foster TC . Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci 1998; 18: 3171–9.
Foster TC . Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Prog Neurobiol 2012; 96: 283–303.
Kumar A, Foster TC . Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol 2004; 91: 2437–2444.
Zanos P, Bhat S, Terrillion CE, Smith RJ, Tonelli LH, Gould TD . Sex-dependent modulation of age-related cognitive decline by the L-type calcium channel gene Cacna1c (Ca v 1.2). Eur J Neurosci 2015; 42: 2499–2507.
Ban TA, Morey L, Aguglia E, Azzarelli O, Balsano F, Marigliano V et al. Nimodipine in the treatment of old age dementias. Prog Neuro Psychopharmacol Biol Psychiat 1990; 14: 525–551.
Riekkinen M, Schmidt B, Kuitunen J, Riekkinen P . Effects of combined chronic nimodipine and acute metrifonate treatment on spatial and avoidance behavior. Eur J Pharmacol 1997; 322: 1–9.
Sandin M, Jasmin S, Levere TE . Aging and cognition: facilitation of recent memory in aged nonhuman primates by nimodipine. Neurobiol Aging 1990; 11: 573–575.
Trompet S, Westendorp RGJ, Kamper AM, de Craen AJM . Use of calcium antagonists and cognitive decline in old age. The Leiden 85-plus study. Neurobiol Aging 2008; 29: 306–308.
Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ et al. Subregion- and cell type-restricted gene knockout in mouse brain. Review. Cell 1996; 87: 1317–26.
Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE et al. Molecular switch from L-type Cav1.3 to Cav1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci 2010; 30: 17051–62.
Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q et al. Cav1.2L-type Ca2+ channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Cav1.3 channels. J Neurosci 2010; 31: 13562–13575.
Terrillion CE, Francis TC, Puche AC, Lobo MK, Gould TD . Decreased nucleus accumbens expression of psychiatric disorder risk gene cacna1c promotes susceptibility to social stress. Int J Neuropsychopharmacol 2017; 20: 428–433, Epub ahead: 1–6.
Roussos P, Mitchell AC, Voloudakis G, Fullard JF, Pothula VM, Tsang J et al. A role for noncoding variation in schizophrenia. Cell Rep 2014; 9: 1417–1429.
Arloth J, Bogdan R, Weber P, Frishman G, Menke A, Wagner KV et al. Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron 2015; 86: 1189–1202.
Bigos KL, Mattay VS, Callicott JH, Straub RE, Vakkalanka R, Kolachana B et al. Genetic variation in Cacna1c affects brain circuitries related to mental illness. Arch Gen Psychiatry 2010; 67: 939–945.
Gershon ES, Grennan K, Busnello J, Badner JA, Ovsiew F, Memon S et al. A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar-associated common SNP of CACNA1C in brain. Mol Psychiatry 2013; 19: 890–894.
Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry 2014; 20: 162–169.
Parkinson Study Group. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease. Mov Disord 2013; 28: 1823–1831.
Acknowledgements
We thank Marcel Schieven for his excellent technical assistance. We thank Günther Schütz for providing Camk2α-CreERT2 mice and Klaus-Armin Nave for providing Nex-Cre mice. This work was partially supported by the German Federal Ministry of Education and Research, within the framework of the e:Med research and funding concept (IntegraMent: Integrated Understanding of Causes and Mechanisms in Mental Disorders; FKZ 01ZX1314H) and by the program for medical genome research within the framework of the NGFN-Plus (FKZ: 01GS08151).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dedic, N., Pöhlmann, M., Richter, J. et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry 23, 533–543 (2018). https://doi.org/10.1038/mp.2017.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.133
This article is cited by
-
Genetic proxies for antihypertensive drugs and mental disorders: Mendelian randomization study in European and East Asian populations
BMC Medicine (2024)
-
A family-based study of genetic and epigenetic effects across multiple neurocognitive, motor, social-cognitive and social-behavioral functions
Behavioral and Brain Functions (2022)
-
Enhanced long-term potentiation and impaired learning in mice lacking alternative exon 33 of CaV1.2 calcium channel
Translational Psychiatry (2022)
-
Phenotypes, mechanisms and therapeutics: insights from bipolar disorder GWAS findings
Molecular Psychiatry (2022)
-
Neuronal deletion of CaV1.2 is associated with sex-specific behavioral phenotypes in mice
Scientific Reports (2022)
