
Abstract
In order to determine the impact of the epigenetic response to traumatic stress on post-traumatic stress disorder (PTSD), this study examined longitudinal changes of genome-wide blood DNA methylation profiles in relation to the development of PTSD symptoms in two prospective military cohorts (one discovery and one replication data set). In the first cohort consisting of male Dutch military servicemen (n=93), the emergence of PTSD symptoms over a deployment period to a combat zone was significantly associated with alterations in DNA methylation levels at 17 genomic positions and 12 genomic regions. Evidence for mediation of the relation between combat trauma and PTSD symptoms by longitudinal changes in DNA methylation was observed at several positions and regions. Bioinformatic analyses of the reported associations identified significant enrichment in several pathways relevant for symptoms of PTSD. Targeted analyses of the significant findings from the discovery sample in an independent prospective cohort of male US marines (n=98) replicated the observed relation between decreases in DNA methylation levels and PTSD symptoms at genomic regions in ZFP57, RNF39 and HIST1H2APS2. Together, our study pinpoints three novel genomic regions where longitudinal decreases in DNA methylation across the period of exposure to combat trauma marks susceptibility for PTSD.
Similar content being viewed by others

Introduction
Exposure to extreme psychological stress such as is experienced during combat situations can result in the development of post-traumatic stress disorder (PTSD).1 Mounting evidence from recent cross-sectional human and animal studies suggests that epigenetic mechanisms, particularly DNA methylation, may play a key role in adaptation to traumatic stress.2, 3, 4, 5 Human studies on the effect of traumatic stress have been constrained by the limited accessibility of the primary tissue type of interest (i.e., the human brain)6 and by difficulties in prospectively capturing the effects of robust and relevant trauma exposures. To investigate the role of DNA methylation following traumatic stress, and its putative role in the development of PTSD, it is useful to focus on longitudinal studies that are less burdened by potential confounders, not the least genetic background. Here, for we believe the first time, a prospective study design in military service members deployed to a combat zone in Afghanistan was used in order to discover differentially methylated positions (DMPs) and differentially methylated regions (DMRs), where longitudinal changes in blood DNA methylation profiles were significantly associated with the development of PTSD. Findings from this methylome discovery study were replicated in an independent prospective military cohort, and were technically validated. Figure 1a and Table 1A illustrate the overall study design.

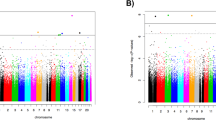
(a) Overall study design. Schematic illustration of the overall study design. Discovery of 17 differentially methylated positions (DMPs) and 12 differentially methylated regions (DMRs) through genome-wide analyses of longitudinal changes in blood-based DNA methylation profiles of military soldiers in relation to the development of psychopathology after deployment to a combat zone was followed by statistical mediation and bioinformatics analyses. Next, these 17 DMPs and 12 DMRs were investigated for replication in an independent prospective military cohort. (b) Manhattan plot of results of PTSD-related differential methylation in the discovery data set. Manhattan plot of the P-values for the regression analyses of longitudinal changes in methylation as a function of changes in PTSD scores in soldiers exposed to traumatic stress during deployment in the discovery data set. Red vertical lines indicate the locations of DMRs. PTSD, post-traumatic stress disorder.
Materials and methods
The overall study design is illustrated in Figure 1a. The study consisted of two different data sets (see Table 1A). The subjects and the methods used for the data sets are indicated below.
Discovery data set
All subjects in the discovery data set were male participants in PRISMO, a large prospective study of 1032 well-characterized Dutch military soldiers scheduled for a deployment of at least 4 months to Afghanistan with longitudinal follow-up.7, 8 This study used data obtained 1 month before and 6 months after the deployment period.
Subject selection
A subset (total n=93) of three similarly sized subgroups of study participants was pre-selected (see also Table 1B) based on the level of traumatic stress exposure and the presence of PTSD symptoms: (i) a subgroup showing high combat-trauma exposure (7.3±2.9) on the deployment experiences checklist, and high levels of post-deployment PTSD symptoms (45.3±8.6) Self-Report Inventory for PTSD; (ii) a subgroup showing high combat-trauma exposure (8.6±2.3) and a low severity of PTSD symptoms (26.0±3.7); and (iii) a subgroup showing low combat-trauma exposure (0.4±0.5) and low levels of post-deployment PTSD symptoms (25.1±3.7). Analysis of age, gender, alcohol consumption, cigarette smoking, military rank, height, weight and medication use did not show any differences among these three groups, although reported exposure to childhood trauma was significantly higher in the group of soldiers who developed symptoms of PTSD after combat trauma (see Table 1B).
Blood samples were collected before and 6 months after deployment. The blood cell-type composition was investigated using flow cytometry, implemented in the clinical laboratory of Utrecht University Medical Center, as previously reported.9 The presence and severity of symptoms of PTSD over the previous 4 weeks were assessed with the 22-item Self-Report Inventory for PTSD (SRIP), which has good reliability and validity. Differences in PTSD symptoms between time points were log-transformed to improve the distribution. Exposure to traumatic stress during deployment was assessed with a 19-item deployment experiences checklist, as previously reported.10 This self-report assessment covered a range of potentially traumatic experiences that occur during deployment, including direct combat stressors. Childhood trauma was assessed with the 27-item Dutch version of the self-reporting short-form Early Trauma Inventory (ETISR-SF),11 assessing early traumatic experiences before the age of 18 years, including general trauma, physical abuse, emotional abuse and sexual abuse. Differences in PTSD symptom scores between time points were log-transformed to improve the distribution. Three missing values in the baseline measure of PTSD symptoms were replaced with the median. General mental health symptoms were also assessed with the Dutch revised version of the Symptom Checklist (SCL-90-R). The SCL-90-R consists of 90 items measuring general mental health, is frequently used in clinical samples and shows good reliability. This study was approved by the ethical committee of University Medical Center Utrecht (01-333/0) and conducted in accordance with the Declaration of Helsinki. All participants gave written informed consent.
Methylome analysis
Genome-wide DNA methylation was assessed using the Illumina Human methylation 450 BeadChip (Illumina, San Diego, CA, USA), which interrogates >485 000 methylation sites per sample at single-nucleotide resolution. Bisulphite-converted DNA was subjected to whole-genome amplification, followed by fragmentation and hybridization of probes. Baseline and follow-up samples were positioned on the same array, and exposure to trauma and PTSD outcomes was equally distributed over the 16 arrays to minimize potential batch effects. Background corrected signal intensities for each probe were extracted using Illumina GenomeStudio software and imported into R. Comparison of non-CpG SNP probes on the array confirmed that two samples per individual were plated on the same array. Probes containing an single-nucleotide polymorphism (SNP) with a minor allele frequency >5% within 10 bp of the single-base extension site according to Illumina’s database were removed from all analyses. Further data quality control and processing steps were conducted using the wateRmelon and SVA packages in R. No sample exhibited 5% of sites with a detection P-value greater than 0.001. A total of 490 sites were removed with beadcount <3 in 5% of samples, and 9728 sites showing 1% of samples with a detection P-value greater than 0.001 were also removed. X-chromosome, Y-chromosome and cross-hybridizing probes probe sequences were also removed.12
Longitudinal changes in DNA methylation per subject were analysed for 39 560 CpG loci. The combat procedure from the SVA package was used to remove plate, sentrix and position effects from the data (Supplementary Figure S1 shows the batch effects before and after removal).
To identify DMPs, longitudinal analyses were conducted using DNA methylation at 6 months after deployment as the outcome via linear regression with DNA methylation levels at 1 month before deployment as a covariate; that is, methylationpostdeployment~ methylation pre-deployment+change in PTSD symptoms. The qq plot of the expected P-values versus the observed P-values and a lambda of 0.998 (see Supplementary Figure S2) indicated absence of type-I error inflation and no artificial differences between groups. False discovery rate (FDR) P-values were calculated according to the Benjamini–Hochberg method. Label-swapping permutations with 100 000 random permuted outcomes were used to derive empirical P-values. The assumptions of the linear regression were evaluated by inspecting the distribution of residuals for the identified loci. The cell-type composition before and after deployment was obtained on the basis of FACS cell counts, as previously described,9 and compared using repeated-measures analysis of variance as well as added to the longitudinal model. The potential influences of cigarette smoking, alcohol use and medication were investigated by excluding participants with documented changes in these parameters from the analysis (N=17, N=5 and N=15, respectively). Gaps in the distribution of the DNA methylation levels were detected with ‘gaphunter, see (http://finzi.psych.upenn.edu/library/minfi/html/gaphunter.html) using a threshold of 0.075 and cutoff value of 0.1. Multilevel analysis, including the band number as random effect, was performed in order to test the potential effect of banding on the associations between longitudinal change in DNA methylation and longitudinal change in PTSD symptoms. For probes showing effects of banding on the longitudinal changes, stratified analyses per band were performed.
To identify DMRs, we used a script that calculated regional P-values using a sliding-window algorithm, as implemented in comb-p code in Python.13 DMRs were defined as three or more adjacent CpG sites on the arrays showing P<0.05. Correction for multiple testing in DMRs was performed using the Šídák FDR at the 0.05% level.
To statistically test for mediation, the standard Baron and Kenny method was used to examine whether the changes in PTSD scores in traumatic stress-exposed subjects are driven by the longitudinal changes in DNA methylation levels at the DMPs and DMRs. First, the direct effect of trauma stress exposure on PTSD scores were tested using multilevel regression model with random intercepts (model 1), and in the second place, the effect of both traumatic stress and DNA methylation were regressed on the phenotype changes (model 2). Regression models were analysed with a linear model ‘lme’ function of nlme package, and the percentage of difference in the coefficient obtained from model 1 and the coefficient obtained from model 2 was calculated. In order to examine whether the difference is significantly different from zero, bootstrapping was applied using boot.ci function from the boot R package. The resulting bootstrap distribution of the changes in the coefficients was plotted and the bias corrected and the accelerated bootstrap confidence interval of the distribution was computed. If zero was not included in the interval, then this was taken as evidence of partial mediation by the methylation changes. Statistical evidence of mediation was concluded if zero was not included in the bootstrap confidence interval, and the percentage of mediation was calculated from the regression models. All statistical analyses were performed using the R language for statistical computing (R version 3.1.2).
Technical validation of the array-based DNA methylation quantifications for the DMRs in ZP57 and RNF39 was performed using pyrosequencing (EpigenDx, Hopkinton, MA, USA; using the Pyrosequencing PSQ96 HS System, Pyrosequencing; Qiagen). RNF39 was measured by sequencing of the region Chr6:30071666-30071627 using a single amplicon that included cg03198009, cg11383134, cg27414868 and cg20228636 but not the CpG cg16078649 that is directly adjacent, but is part of the original DMR. The ZFP59 DMR was measured using two amplicons (Chr6:29680852-29680731) that included all CpGs from the ZFP59 DMR (cg10568066, cg12633154, cg13401893 and cg09279736). The average methylation level for the CpG sites was calculated for the two DMRs as were parametric correlations with the array-based data.
Bioinformatic analyses
The numbers of CpGs probes per gene were first corrected using a logistic modelling approach, based on an algorithm previously published.14 Gene Ontology (GO) analysis was then performed using GOrilla to identify overrepresented GO terms at the 0.05 Šídák-adjusted level, based on genes showing changes in DNA methylation for the Molecular Function, Cellular Component and Biological Process ontologies. Pathway analysis was performed using PathVisio 3 with the human WikiPathways analysis collection (downloaded 12 March 2015). To map data points to pathway elements, the identifier mapping database Hs_Derby_20130701.bridge was used. Through overrepresentation analysis, pathways that contained a higher than expected number of genes with altered methylation were identified. In order to filter GO terms with overlapping genes, an additional logistic model was run on all GO terms containing 10 to 2000 genes and having a one-sided P-value of 0.05 (Z-score 1.645).
For the GO and pathway analyses, the most significant CpG according to GREAT annotation was selected for each gene, yielding a data set containing 15 950 unique genes, among which 6774 mapped to at least one pathway in the collection. Genes with a DMP showing an absolute methylation change of at least 0.01 and a maximum Šidák corrected P-value of 0.05 for their most significant CpG island were considered as significantly changed.
Network analysis was performed based on the genes associated with the DMRs. Genes near or consisting the DMRs (rather than the DMPs) were used for the network analysis because DMRs reflect more robust genomic alterations in DNA methylation than DMPs and because the DMR analysis yielded an appropriate numbers of genes for performing network analyses. An initial network was constructed by running GeneMania using the default of adding 20 extra nodes based on a variety of sources. The resulting network was imported into Cytoscape 3, which was used to lay out the network and map the data (methylation changes and Šídák-adjusted P-values) to the nodes.
Replication data set
Replication data were obtained from the Marine Resiliency Study, a prospective PTSD study with longitudinal follow-up (pre- and post-exposure to combat stress) of 2,599 US Marines bound for deployment to Iraq/Afghanistan.15 Baseline measures were recorded approximately one month pre-deployment. Follow-up was performed at approximately 1 week, 3 months and six months post-deployment. Post-traumatic stress (PTS) symptoms were assessed using a structured diagnostic interview and the Clinician Administered PTSD Scale (CAPS), and combat exposure was assessed using the Deployment Risk and Resilience Inventory (DRRI).16 A homogeneous subset of 98 men from the MRS was selected for DNA methylation analyses. The mean combat exposure score using the DRRI for these subjects was 0.77 (s.d. 0.62). The selected subjects showed no PTSD diagnosis (CAPS⩽25) before deployment and reported relatively high combat exposure during the index deployment. At six months post-deployment, 35 cases were diagnosed with partial or full PTSD with a mean CAPS score of 55.03 (standard deviation 16.05), and 63 controls showed no PTSD symptoms at any time point (CAPS⩽25). The institutional review boards of the University of California San Diego, VA San Diego Research Service, and Naval Health Research Center approved the study. All subjects provided informed consent.
Genome-wide DNA methylation levels were assessed in DNA extracted from whole blood using the Infinium HumanMethylation450 array. Baseline and follow-up samples were positioned differently between studies. Methylation level βs were calculated using the Minfi package and normalized to correct for type-I and II probe design bias using the BMIQ procedure implemented in watermelon. Batch and plate effects were removed using COMBAT. Relative proportions of cell compositions were estimated to account for cellular heterogeneity in blood-derived samples using the Minfi package.17 Analyses were targeted at the DMPs and DMRs identified in the discovery.
Relevance beyond PTSD
For the DMP and DMRs discovered in the Dutch military cohort and subsequently replicated, we also explored whether changes in DNA methylation levels were related to global expression of symptoms of mental disorders; broader than PTSD. Global expression of symptoms of mental disorders was assessed using the Symptom CheckList 90 (SCL-90) questionnaire at baseline and the 6 months follow-up within the Dutch military cohort. Regressions analyses tested for associations between longitudinal changes in DNA methylation and longitudinal changes in SCL-90 scores.
Results
Discovery of PTSD susceptibility in a Dutch military cohort: DMPs
After FDR adjustment at the 0.05 level, regression analyses identified 17 genome-wide significant CpG sites (DMPs) in/near the genes DUSP22, NINJ2, HOOK2, PAX8, COL1A2, SDK1, MYT1L and HIST1H2APS2, in which longitudinal changes in DNA methylation were associated with increasing PTSD symptoms (Figure 1b and Table 2A). The direction of effect for all identified DMPs was negative, meaning that increases in PTSD symptoms were associated with decreases in DNA methylation at those DMPs over time.
To statistically test whether DNA methylation at the identified CpG sites may be a causal mediator in the association between traumatic stress exposure and longitudinal changes in PTSD symptoms, bootstrapping analyses (N=10 000) were performed in the trauma-exposed subjects showing increased PTSD symptoms over time. These analyses indicated that the strength of the association between traumatic stress exposure and the longitudinal changes in PTSD symptom scores was co-dependent on the methylation of three DMPs in HOOK2 and 1 DMP in MYT1L (see Table 2A). The DNA methylation changes observed at the DMPs accounted for a significant but very small proportion of the variance in the longitudinal changes in PTSD symptoms, that is, the individual DMPs explained between 0.4 and 0.7% of the variance (see Table 2A), while a conjunct analysis of the seven genome-wide DMPs showing statistical mediation indicated that these DMPs together explained 1.3% of the variance.
Discovery of PTSD susceptibility: DMRs
Twelve DMRs (see Table 2B) were identified, where longitudinal changes in average DNA methylation were associated with longitudinal changes in PTSD symptoms. These DMRs were located in or near the transcription start sites of several genes. The direction of the association with PTSD symptoms was negative for all loci in the DMRs, indicating that increased PTSD symptom scores over time were associated with decreased DNA methylation levels at the DMRs.
The strength of the observed associations between traumatic stress and PTSD symptoms was mediated by DNA methylation changes of seven DMRs in HOOK2, PAX8, SPATC1L, PM20D1, SMAD5, GPR12 and RNF39 (see Table 2B) although mediation accounted for a very small proportion (between 0.1 and 0.8%) of the variance in changes in PTSD symptoms (see Table 2B).
The results of sensitivity analyses for the potential confounders childhood trauma, age, actual time in weeks between baseline and follow-up measurements, cell-type-composition, or changes in smoking, medication and alcohol use indicated no significant effects of childhood trauma levels, age, actual time in weeks between baseline and follow-up measurements, cell-type composition and change in use of alcohol or medication, while change in smoking influenced the association between DNA methylation changes of the DMR in SMAD5, but not for any other DMP or DMR.
Although probes containing an SNP with a minor allele frequency >5% and located within 10 bp of the single-base extension site according to Illumina’s database were removed in the analysis pipeline, banding effects of the methylation levels remained for several loci. Despite the fact that banding is not likely to influence the results due to the longitudinal nature of the analysis, we further investigated the potential impact. For that purpose we used the ‘gaphunter’ function from the minfi package17 to identify possible banding of the DNA methylation levels at the DMPs and DMRs. Distinct bands of methylation levels were identified in 12 of the 17 DMPs and in 5 of the 12 DMRs (see Tables 2A and 2B). The proportion of CpG sites showing banding was much higher in the DMPs than expected by chance (χ2=1 299 628, df=3, P-value<2.2e-16). As the bands were suggestive of genetic effects in cis or trans, previously published mQTL data sets were explored and all bands could be connected with mQTLs, thus suggesting that genetic effects underlie the banding in distribution of DNA methylation levels. Next, the influences of the bands per locus on the results of longitudinal analyses were investigated using multilevel analysis using the bands as random factor in a mixed effect model. The identified bands did not influence the observed associations between longitudinal changes in PTSD symptoms and changes in DNA methylation for 16 out of the 17 DMPs and at 10 out of the 12 DMRs. Multilevel analysis however rendered the associations for the DMP cg04657146 in HOOK2, the DMR in HOOK2 and the DMR in SMAD5 (see Tables 2A and 2B) nonsignificant. Stratified analyses per band for these loci showed that the significant associations were retained only for distinct bands in the DMP at cg04657146 in HOOK2 and in the DMRs in HOOK2 and SMAD5. Thus, these results suggest interplay between genetic background and DNA methylation changes in the development of PTSD, particularly for these loci.
DNA methylation marks and traumatic stress
Inclusion of a deployed but unexposed control group in this prospective study enabled us to separately investigate associations between the severity of traumatic stress exposure during deployment and longitudinal changes in DNA methylation in subjects who did not develop PTSD symptoms after deployment (i.e. resilient individuals). These analyses were performed in individuals who showed no significant changes in PTSD symptom scores over time but reported either low or high levels of traumatic stress (see Table 1B). The analyses were constrained to CpG sites in the 17 DMPs and 12 DMRs exhibiting a significant genome-wide association with changes in PTSD symptoms. The results indicated that exposure to traumatic stress in these individuals was associated with longitudinal increases (rather than decreases) in DNA methylation at the DMPs and DMRs (see Figure 2). In contrast to the negative associations between PTSD symptoms and DMPs and DMRs observed in the trauma-exposed groups (see Figure 2 and, for the network between the DMRs, Figure 2a), trauma exposure was positively associated with changes in DNA methylation levels at these DMPs and DMRs in military subjects who did not show increases in PTSD symptoms but were differentially exposed to traumatic stress (see Figure 2b). In other words, whereas trauma exposure increased DNA methylation levels at the DMPs and DMRs over time in the group that did not develop PTSD symptoms, the group that developed PTSD symptoms after trauma exposure showed decreased DNA methylation at the identified DMPs and DMRs (see also Supplementary Figure S3 which exemplifies the results on the DMRs in ZFP57 and RNF39). These results reveal opposite effects in susceptibility and resilience to the effects of traumatic stress exposure, which are thus related to directionally differential DNA methylation responses, at least for the investigated DMPs and DMRs.

Boxplot of the results of pathway analyses. Pathways containing more than the expected number of genes with PTSD-associated changes in DNA methylation levels. The data were imported into PathVisio, corrected for the number of probes per gene and overrepresentation analysis of the WikiPathways curated for analysis pathway collection was performed using a significant Šídák-adjusted P-value of <0.05 and a change in the methylation estimate of at least 0.01 as criteria. Z-scores of significant pathways (Z-score>1.96) are displayed. Full tables of the pathway overrepresentation analysis results are provided in Supplementary Tables S4 and S5. PTSD, post-traumatic stress disorder.
The results of the bioinformatics analyses of gene ontology and pathway enrichment as well as network analyses are described in the Supplementary Tables S1 and S2, Supplementary Figure S4 and summarized in Figure 3. The analyses identified significant enrichment in a range of pathways (see Figure 2 and Supplementary Tables S1 and S2) including several pathways with clear relevance for brain functioning and stress-related disorders, such as ‘Sudden Infant Death Syndrome (SIDS) Susceptibility Pathways’, ‘Circadian rhythm related genes’, ‘Wnt Signaling Pathway and Pluripotency’, ‘Dopaminergic neurogenesis’, ‘Neural Crest Differentiation’, ‘IL17 signaling pathway’, ‘TSH signaling pathway’, ‘Serotonin Receptor 2 and ELK-SRF/GATA4 signaling’, and ‘Serotonin Transporter Activity’.

Network of genetically annotated regions showing differential DNA methylation. Networks of genes in differentially methylated regions (DMRs) and their direct interaction partners. Genes in/near DMRs (Table 2B) were uploaded into GeneMania. Interaction partners of several types were added, using the default settings, extending the network with 20 additional nodes (interaction partners). Three genes were not connected (PM20D1, WASF3, ZFP57). The resulting network was imported into Cytoscape. The data were mapped to the network, visualizing the estimated methylation changes associated with increased PTSD symptom scores in subjects exposed to traumatic stress (a) and with exposure to trauma in subjects without increased PTSD symptom scores (b) using a colour gradient. The border size represents the P-value: the Šídák-adjusted P-value for the DMR for genes in DMRs (both panels) and, for others, the most significant individual Šídák-adjusted P-value for all associated CpGs according to GREAT annotation (a). Genes with a transcriptional start site near the DMRs are displayed as rounded boxes, and partners added by GeneMania are displayed as ellipses. Edges are visualized according to the GeneMania colour scheme. The data for the X-chromosomal XIAP were not available in the data set. Two DMRs were located near the transcription start site of DUSP22, and the most significant P-value for these two is shown. PTSD, post-traumatic stress disorder.
Replication of DMRs and DMPs related to PTSD susceptibility
The 17 DMPs and 12 DMRs that were identified in the discovery data set were tested in the replication data set using the same regression analysis of longitudinal changes in DNA methylation, in which longitudinal changes in PTSD symptom scores were the main determinant of longitudinal changes in DNA methylation. In addition, the data were adjusted for three ancestry-related principal components to deal with ethnic diversity in this sample.18 Replication was obtained for two DMRs and one DMP at which longitudinal changes in PTSD symptom scores were significantly associated (with the same directionality) with changes in DNA methylation (see also Table 2A). The two replicated DMRs were the DMR in ZFP57 at chromosome six between positions 29 648 161 and 29 649 025 (near the transcription start site of ZFP57; P=1.32E-14) and the DMR in RNF39 between positions 30 039 374 and 30 039 601 (near the transcriptional start site of RNF39 and PPP1R11; P=0.0026) at chromosome six. The replicated DMP was located in HIST1H2APS2 (cg03517284 B=−0.15, P=0.002). Although these showed the same direction of effect in the replication data as in the discovery, we also observed that the DMPs located in DUSP22 (cg03395511 B=0.44, P=0.005; cg18110333 B=0.59, P=0.016; cg21548813 B=0.38, P=0.028; cg01516881 B=0.21, P=0.027) and SDK1 (cg07249765 B=0.50, P=0.017) showed a similar statistical association between changes in DNA methylation and changes in PTSD status in the replication data, but with opposite directionality as in the discovery data set (see also Table 2A). Therefore, DUSP22 and SDK1 methylation changes were considered to be non-replications. A limitation of the replication was that the distribution of the baseline and follow-up samples across arrays and positions was different and differences between batch correction procedures may have influenced the robustness of replications attempt. Sensitivity analyses adjusting for changes in the cell-type distribution did not alter the significance of replicated findings.
Technical validation
The robustness of the array-based DNA methylation data was validated by pyrosequencing the same CpG sites of DMRs in ZFP57 and RNF39 in the discovery data set. High correlations were observed between pyrosequencing-based DNA methylation results and the array-based results for the average DNA methylation level of the loci in the ZFP57 DMR (correlation=0.96, P<0.001) and for the average DNA methylation level of the loci in the RNF39 DMR (correlation=0.95, P<0.001) (see also Supplementary Figure S5). The associations between longitudinal alterations in PTSD symptom scores and longitudinal alterations in the average DNA methylation level remained statistically significant for the DMR in ZFP57 (B=−2.49, P=0.021) when using pyrosequencing-based data, although this was not observed for the pyrosequencing-based data for the average DNA methylation levels of the DMR in RNF39 (B=−1.12, P=0.36).
Associations with broader expression of psychopathology
Longitudinal changes in scores on the SCL-90 questionnaire in the discovery data set were significantly associated with longitudinal changes in DNA methylation levels in the DMP in HIST1H2APS2 (cg03517284 B=−0.05, P=0.033) and the DMR in ZFP57 (B=−0.06, P=0.008) but not with the DMR in RNF39 (B=−0.03, P=0.346). The main results on associations with the DMRs in ZFP57 and RNF39 and the DMP in HIST1H2APS2 are summarized in Table 3.
Discussion
This is the first hypothesis-free study of longitudinal changes in blood DNA methylation profiles across the period of exposure to combat trauma in relation to the development of PTSD. Genome-wide discovery with targeted replication of within-subject alterations in DNA methylation revealed that decreasing DNA methylation levels at the genes ZFP57, RNF39 and HIST1H2APS2 (all located on chromosome six) were related to the increasing levels of PTSD symptoms over time. Bioinformatics analyses of the identified genes also indicated significant enrichment for the neuronal compartment in important brain-related biological pathways.
A major strength of the present study is the prospective sampling and phenotype assessments conducted before and after a period of deployment in subgroups enriched for trauma exposure and PTSD outcomes for genome-wide discovery with targeted replication in a second military prospective military cohort. The main findings were technically validated using bisulphite pyrosequencing, and although the analytical sensitivity of bisulfite pyrosequencing is ~5–10% for individual CpG sites and, thus, does not have the sensitivity necessary to detect small (i.e. <5%) changes in DNA methylation,19 we still observed evidence for the robustness of associations when using the pyrosequencing-based data. The longitudinal design of our study furthermore reduced the likelihood of confounding, including confounding by the genetic variants eliminating both cis and trans effects of the genotype on DNA methylation, as has previously been observed.20, 21, 22
The strongest evidence was found for two DMRs, one in ZFP57 and one in RNF39. Susceptibility to PTSD and broader expression of psychopathology were associated with hypomethylation at ZFP57 in two independent samples, and by using two techniques for measuring DNA methylation levels. ZFP57 encodes zinc-finger protein 57, which functions as a transcriptional regulator of genomic imprinting in stem cells during development and in adult life.23, 24 ZFP57-dependent genomic imprinting requires the co-factor KAP1, and a recent rodent study has observed that conditional knock-out of KAP1 in the hippocampus induces behavioural susceptibility to stress, and is associated with epigenetic marks on a distinct set of genes in the hippocampus.25 Thus, together, these data are consistent with a key role for ZFP57-KAP1 genomic imprinting in susceptibility to traumatic stress. This role in imprinting raises the question of whether this locus is involved in transgenerational inheritance of disease vulnerability.
The second DMR was located in RNF39. We discovered and replicated associations between susceptibility to develop PTSD upon traumatic stress exposure and hypomethylation in the RNF39 DMR in the blood. DNA methylation at RNF39 significantly mediated the associations between traumatic stress exposure and changes in PTSD symptoms. These findings suggest that RNF39 is a key gene regulator of stress susceptibility in humans, a finding in line with results from genome-wide association studies showing that common variants in RNF39 increase the risk of schizophrenia,26 anger27 and risk of major psychiatric disorders.28 RNF39 encodes ring finger protein 39, which plays a role in the early phase of synaptic plasticity and in long-term potentiation.29 A recent micro-array study has shown that rodents previously exposed to stress show elevated RNF39 mRNA expression in the hippocampal dentate gyrus in response to corticosterone challenge, as compared with animals not exposed to prior stress.30 Another animal stress study has shown that RNF39 mRNA levels are also strongly upregulated in the lateral amygdala during Pavlovian fear conditioning.31 Together, these findings support the role of RNF39 in mediating the effect of stress on central fear and stress circuitries in the brain and on stress-related behaviour.
It is noteworthy that replication of the DMRs was more successful compared with that of the DMPs. Two DMRs and only one DMP showed consistent effects across data sets, and can thus be considered as candidate regions for susceptibility to trauma-related disorder. It also suggests a superior biological meaning of DMRs and lower rate of false positives for such regions as compared with single CpG loci.
The analysis of longitudinal DNA methylation changes in the discovery data set indicated opposite directions of the relationship with trauma compared with PTSD symptom levels in all genome-wide significant DMPs and DMRs. This divergence in the general response to trauma and the development of symptoms is consistent with a maladaptive response to trauma in those who develop PTSD and raises the question of whether the differential psychological and behavioural responses to traumatic stress may be traced to the epigenomic level. The mechanisms underlying these epigenetic changes remain to be established but may include inter-individual differences in one-carbon metabolism, including folate levels or a number of other regulators of transcription.
The bioinformatic pathway analyses of the discovery data set further showed strong links between the development of symptoms of PTSD and DNA methylation in distinct biological pathways with relevance for neuronal functioning and with prior evidence for mediation of stress susceptibility and in PTSD. These pathways comprised the dopaminergic and serotonergic neurotransmission,32, 33 WNT signalling pathway and β-catenin;34 the hypothalamus–pituitary–adrenal axis, including CRF signalling;4, 35 circadian rhythm, complement activation, the immune system including IL-17 signalling; and the brain-derived neurotrophic factor and neurotrophin signalling pathways.
The results of this study should be interpreted in the context of several limitations. Further studies of the identified loci and genes are needed to establish the biological relevance in the brain in relation to trauma and trauma-related mental disorders. The limitations include the relatively small sample size and the use of a male, mainly Caucasian population, thus precluding generalization of our findings to females, other trauma types and other populations. Indeed, the limited replication also suggests that differences between studies (and study designs) are large, and many confounders may be at play including, for example, diet and immune status. In addition to population-specific properties, the observed DNA methylation changes were small, that is, lower than 5%, which may have influenced the technical validation of array-based and pyrosequencing-based DNA methylation analyse (particularly for loci with low DNA methylation level, e.g., the RNF39 DMR). Thus, initiatives to converge epigenetic studies using DNA samples collected in large cohorts of patients with PTSD and trauma-exposed controls,36 employed with similar epidemiological designs and analysed simultaneously in the same or parallel batches of arrays, are warranted in order to reduce false-positive findings and optimize the power of these discovery endeavours.
Overall, this study is effective in highlighting those loci that have strong and consistent association with disease risk. Identification of these loci thereby provides new leads for the understanding of the mechanisms of differential responses to traumatic stress and for how epigenetic alterations may play a role in the development of PTSD.
References
Yehuda R, Hoge CW, McFarlane AC, Vermetten E, Lanius RA, Nievergelt CM et al. Post-traumatic stress disorder. Nat Rev Dis Primers 2015; 1: 15057.
Zannas AS, Provencal N, Binder EB . Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry 2015; 78: 327–335.
Vinkers CH, Kalafateli AL, Rutten BP, Kas MJ, Kaminsky Z, Turner JD et al. Traumatic stress and human DNA methylation: a critical review. Epigenomics 2015; 7: 593–608.
Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA 2013; 110: 8302–8307.
Uddin M, Galea S, Chang SC, Aiello AE, Wildman DE, de los Santos R et al. Gene expression and methylation signatures of MAN2C1 are associated with PTSD. Dis Markers 2011; 30: 111–121.
Tsankova N, Renthal W, Kumar A, Nestler EJ . Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007; 8: 355–367.
Reijnen A, Rademaker AR, Vermetten E, Geuze E . Prevalence of mental health symptoms in Dutch military personnel returning from deployment to Afghanistan: a 2-year longitudinal analysis. Eur Psychiatry 2015; 30: 341–346.
Eekhout I, Reijnen A, Vermetten E, Geuze E . Post-traumatic stress symptoms 5 years after military deployment to Afghanistan: an observational cohort study. Lancet Psychiatry 2015; 3: 58–64.
Boks MP, Mierlo HC, Rutten BP, Radstake TR, De Witte L, Geuze E et al. Longitudinal changes of telomere length and epigenetic age related to traumatic stress and post-traumatic stress disorder. Psychoneuroendocrinology 2014; 51: 506–512.
van Zuiden M, Geuze E, Willemen HL, Vermetten E, Maas M, Heijnen CJ et al. Pre-existing high glucocorticoid receptor number predicting development of posttraumatic stress symptoms after military deployment. Am J Psychiatry 2011; 168: 89–96.
Vermetten E, Bremner JD, Skelton L, Spiegel D . PTSD and Vietnam Veterans. Science 2007; 315: 184–187; author reply 184–187.
Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8: 203–209.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ . Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012; 28: 2986–2988.
Lunnon K, Hannon E, Smith RG, Dempster E, Wong C, Burrage J et al. Variation in 5-hydroxymethylcytosine across human cortex and cerebellum. Genome Biol 2016; 17: 27.
Baker DG, Nash WP, Litz BT, Geyer MA, Risbrough VB, Nievergelt CM et al. Predictors of risk and resilience for posttraumatic stress disorder among ground combat Marines: methods of the Marine Resiliency Study. Prev Chronic Dis 2012; 9: E97.
Minassian A, Maihofer AX, Baker DG, Nievergelt CM, Geyer MA, Risbrough VB et al. Association of predeployment heart rate variability with risk of postdeployment posttraumatic stress disorder in active-duty marines. JAMA Psychiatry 2015; 72: 979–986.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30: 1363–1369.
Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, Miller MW et al. Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: a genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology 2015; 51: 459–471.
Mikeska T, Craig JM . DNA methylation biomarkers: cancer and beyond. Genes (Basel) 2014; 5: 821–864.
Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 2009; 4: e6767.
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 2016; 19: 40–47.
Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci 2016; 19: 48–54.
Plant K, Fairfax BP, Makino S, Vandiedonck C, Radhakrishnan J, Knight JC . Fine mapping genetic determinants of the highly variably expressed MHC gene ZFP57. Eur J Hum Genet 2014; 22: 568–571.
Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 2008; 15: 547–557.
Jakobsson J, Cordero MI, Bisaz R, Groner AC, Busskamp V, Bensadoun JC et al. KAP1-mediated epigenetic repression in the forebrain modulates behavioral vulnerability to stress. Neuron 2008; 60: 818–831.
Sullivan PF, Lin D, Tzeng JY, van den Oord E, Perkins D, Stroup TS et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry 2008; 13: 570–584.
Mick E, McGough J, Deutsch CK, Frazier JA, Kennedy D, Goldberg RJ . Genome-wide association study of proneness to anger. PLoS ONE 2014; 9: e87257.
Lotan A, Fenckova M, Bralten J, Alttoa A, Dixson L, Williams RW et al. Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Front Neurosci 2014; 8: 331.
Matsuo R, Asada A, Fujitani K, Inokuchi K . LIRF, a gene induced during hippocampal long-term potentiation as an immediate-early gene, encodes a novel RING finger protein. Biochem Biophys Res Commun 2001; 289: 479–484.
Datson NA, van den Oever JM, Korobko OB, Magarinos AM, de Kloet ER, McEwen BS . Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology 2013; 154: 3261–3272.
Ploski JE, Park KW, Ping J, Monsey MS, Schafe GE . Identification of plasticity-associated genes regulated by Pavlovian fear conditioning in the lateral amygdala. J Neurochem 2010; 112: 636–650.
Feder A, Nestler EJ, Charney DS . Psychobiology and molecular genetics of resilience. Nat Rev Neurosci 2009; 10: 446–457.
LaPlant Q, Vialou V, Covington HE 3rd, Dumitriu D, Feng J, Warren BL et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci 2010; 13: 1137–1143.
Dias C, Feng J, Sun H, Shao NY, Mazei-Robison MS, Damez-Werno D et al. beta-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature 2014; 516: 51–55.
Daskalakis NP, Cohen H, Cai G, Buxbaum JD, Yehuda R . Expression profiling associates blood and brain glucocorticoid receptor signaling with trauma-related individual differences in both sexes. Proc Natl Acad Sci USA 2014; 111: 13529–13534.
Logue MW, Amstadter AB, Baker DG, Duncan L, Koenen KC, Liberzon I et al. The Psychiatric Genomics Consortium Posttraumatic Stress Disorder Workgroup: posttraumatic stress disorder enters the age of large-scale genomic collaboration. Neuropsychopharmacology 2015; 40: 2287–2297.
Acknowledgements
The recruitments and assessments and subjects in the discovery data set were funded by the Dutch Ministry of Defence, and the DNA methylation and mRNA analyses of discovery data set were funded by the VENI Award fellowship from the Netherlands Organisation for Scientific Research (NWO, grant number 916.11.086) to BPFR. Statistical analyses were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara and financially supported by the Netherlands Scientific Organization (NWO 480-05-003 PI: Posthuma) along with a supplement from the Dutch Brain Foundation and VU University Amsterdam. The Marine Resilience Study was funded by the United States Department of Veterans Affairs Health Service Research and Development project SDR 09-0128, the Marine Corps, and the Navy Bureau of Medicine and Surgery (to DGB) and NIH 1 R01MH093500 (to CN).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Disclosure
The funders had no role in the design and reporting of the study.
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Rutten, B., Vermetten, E., Vinkers, C. et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol Psychiatry 23, 1145–1156 (2018). https://doi.org/10.1038/mp.2017.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.120
This article is cited by
-
Differential methylation of linoleic acid pathway genes is associated with PTSD symptoms – a longitudinal study with Burundian soldiers returning from a war zone
Translational Psychiatry (2024)
-
Prominent genetic variants and epigenetic changes in post-traumatic stress disorder among combat veterans
Molecular Biology Reports (2024)
-
Transcription and DNA methylation signatures of paternal behavior in hippocampal dentate gyrus of prairie voles
Scientific Reports (2023)
-
Epigenetics of early-life adversity in youth: cross-sectional and longitudinal associations
Clinical Epigenetics (2022)
-
An epigenetic association analysis of childhood trauma in psychosis reveals possible overlap with methylation changes associated with PTSD
Translational Psychiatry (2022)
