
Abstract
The lack of strong association between brain beta-amyloid deposition and cognitive impairment has been a challenge for the Alzheimer’s disease (AD) field. Although beta-amyloid is necessary for the pathologic diagnosis of AD, it is not sufficient to make the pathologic diagnosis or cause dementia. We sought to identify the genetic modifiers of the relation between cortical beta-amyloid burden (measured using [18F]Florbetapir-PET) and cognitive dysfunction (measured using ADAS-cog) by conducting a genome-wide interaction study on baseline data from participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) phases GO/2 (n=678). Near genome-wide significant interaction effect was observed for rs73069071 within the IAPP (amylin) and SLCO1A2 genes (P=6.2 × 10−8). Congruent results were found using data from participants followed up from ADNI-1 (Pone-tailed=0.028, n=165). Meta-analysis across ADNI-GO/2 and ADNI-1 revealed a genome-wide significant interaction effect (P=1.1 × 10−8). Our results were further supported by similar interaction effects on temporal lobe cortical thickness (whole-brain voxelwise analysis: familywise error corrected P=0.013) and longitudinal changes in ADAS-cog score and left middle temporal thickness and amygdalar volume (Pone-tailed=0.026, 0.019 and 0.003, respectively). Using postmortem beta-amyloid immunohistochemistry data from 243 AD participants in the Religious Orders Study and Memory and Aging Project, we also observed similar rs73069071-by-beta-amyloid deposition interaction effect on global cognitive function (Pone-tailed=0.005). Our findings provide insight into the complexity of the relationship between beta-amyloid burden and AD-related cognitive impairment. Although functional studies are required to elucidate the role of rs73069071 in AD pathophysiology, our results support the recently growing evidence on the role of amylin in AD.
Similar content being viewed by others
Introduction
Beta-amyloid (Aβ) formation is a histopathological hallmark of Alzheimer’s disease (AD).1 Accumulation of Aβ fibrils is an early event in the pathological cascade of AD which occurs before structural brain atrophy and cognitive decline.1 The ‘amyloid hypothesis’ that considers Aβ as a causal factor in AD is well supported by rare early-onset autosomal dominant cases of AD.2 However, the mechanism by which Aβ contributes to late-onset sporadic AD is less clear and likely more complex.3 Although Aβ deposition is necessary for the pathologic diagnosis of AD, it is not sufficient in and of itself to cause cognitive dysfunction and clinical dementia. Substantial overlap is observed in the amount of Aβ deposition across the continuous spectrum of cognitive impairment. Various studies have identified individuals with high Aβ burden with no or minimal cognitive deficits,4 whereas others have shown that high Aβ deposition has low specificity for predicting development of AD dementia.5, 6 In addition, many studies have demonstrated weak to moderate associations between amyloid burden and cognitive dysfunction.4, 7, 8, 9, 10
Recent advances in the development of positron emission tomography (PET) radiotracers made it possible to quantify brain Aβ deposition non-invasively.11 Aβ-PET can have an important role in determining the mechanisms underlying the susceptibility or resistance to AD and its progression in human cohorts. In this study, using data from participants originally enrolled in the Alzheimer’s Disease Neuroimaging Initiative (ADNI)12 phases GO/2, we conducted a genome-wide interaction analysis to identify the genetic variants that modify the relationship between cortical Aβ deposition and cognitive dysfunction. Next, we attempted to replicate our findings in the data from participants followed up from ADNI-1. We further investigated how the identified genetic variants alter the relationship between baseline Aβ deposition and cortical thickness and also longitudinal changes in cognitive and atrophy measures. Finally, using data from the Religious Orders Study and Rush Memory and Aging Project (ROS/MAP)13, 14 we sought to replicate our findings in this independent postmortem AD neuropathology sample. We hypothesized that identification of genetic variants that modulate the impact of Aβ deposition on cognitive performance could help clarify Aβ-related molecular processes associated with cognitive impairment in late-onset AD.
Materials and methods
Participants were healthy elderly and mild cognitive impairment (MCI) and AD patients from ADNI and ROS/MAP cohorts. All ADNI participants provided written informed consent, and the institutional review board of each ADNI site approved study protocols. All ROS/MAP participants signed an informed consent and Anatomical Gift Act, and ROS and MAP studies were approved by the institutional review board of Rush University Medical Center. Methods are described in detail in Supplementary Methods.
ADNI data
Cortical Aβ deposition (average uptake of frontal, anterior/posterior cingulate, lateral parietal and lateral temporal cortical regions standardized to uptake in the cerebellum) was estimated using [18F]Florbetapir-PET.15 Cognitive dysfunction was evaluated using the Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog), which consists of 11 tasks in cognitive domains mainly consisting of memory, language and praxis,16 and verbal memory performance was assessed using Rey Auditory Verbal Learning Test (RAVLT).17 T1-weighted brain magnetic resonance imaging (MRI) was performed on 3.0T MR scanners. Cerebrospinal fluid (CSF) Aβ1–42 levels were also measured.18 Genotyping was performed using the Illumina HumanOmniExpress BeadChip in ADNI-GO/2 and using the Illumina Human610-Quad BeadChip in ADNI-1. Given the differences in genotyping kits and the fact that ADNI-1 participants had underwent cognitive assessment at multiple time points prior to their PET scan, data from ADNI-1 were analyzed separately as a replication sample (Table 1).
We imputed the genetic ADNI-GO/2 and ADNI-1 data separately after quality control. The genome-wide interaction study and all further statistical analyses were conducted using imputed data from participants with European ancestry while controlling for the effects of age, sex and education years and assuming an additive mode of action for genetic variants, unless otherwise specified.
After structural MRI preprocessing, voxel-based non-parametric statistical analysis was performed on cortical thickness images to evaluate the genotype-by-cortical Aβ deposition interaction effect on regional cortical thickness, while controlling for the effects of age, sex, handedness and education years.
Longitudinal analyses were performed on available ADNI-GO/2 cognitive and structural MRI longitudinal data using linear mixed-effects models assuming a random intercept and slope per individual.
ROS/MAP data
Global cognition scores were computed as the average of the normalized Z-scores of 17 tasks in 5 cognitive domains (episodic, semantic and working memory, and perceptual orientation and speed).19 Quantification of Aβ deposition was accomplished using immunohistochemistry and automated image processing of tissue samples from hippocampus (CA1/subiculum), angular gyrus, and entorhinal, superior frontal, dorsolateral prefrontal, inferior temporal, anterior cingulate and occipital (calcarine) cortices.20 Using the modified Bielschowsky silver-staining technique, neuritic and diffuse plaques were visualized in tissue sections from hippocampus, and midfrontal, superior/middle temporal, inferior parietal and entorhinal cortices.21 Quantitative composite scores were computed separately for overall Aβ burden (mean percent area occupied by Aβ across regions) and neuritic and diffuse plaques (average standardized regional density) for each individual as previously described.20, 21 Self-declared non-Hispanic Caucasian participants were genotyped using the Affymetrix GeneChip 6.0 platform and their genomic data were imputed22 after quality control.
Analyses were performed on latest available data from participants whose last antemortem cognitive assessment was performed within 3 years of the time of death (Table 1). All statistical analyses were conducted while controlling for the effects of age at last cognitive assessment, sex, education years and study (ROS vs MAP).
Statistical analysis
Reported P-values are two-tailed, unless otherwise specified. For analyses in the replication sets and secondary analyses, one-tailed P-values are reported given the expectation for effects in the same direction with results from the discovery set (that is, cross-sectional ADNI-GO/2 data), as per other genome-wide association studies.23, 24, 25, 26, 27
Results
Genome-wide interaction study
Data from 678 participants originally enrolled in ADNI-GO/2 were included in the genome-wide single-nucleotide polymorphism (SNP)-by-cortical Aβ deposition interaction study in relation to performance in ADAS-cog. Near genome-wide significant interaction effects were observed for 7 imputed variants located on chromosome 12p12.1 (peak-P=6.2 × 10−8; IMPUTE2 info score>0.99) and another imputed variant (rs112821268; IMPUTE2 info score=0.68) located on chromosome 13q31.1 (P=7.8 × 10−8). As the 7 variants on chromosome 12 were in high linkage disequilibrium (LD: r2>0.99), we selected one of them with the peak P-value (rs73069071) for further analyses. No other SNP included in the genome-wide study showed P<1 × 10−7 (Figure 1).

Results of genome-wide SNP-by-cortical Aβ deposition (as measured by [18F]Florbetapir-PET) interaction study in relation to Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog), while accounting for the effects of age, sex and years of education, using baseline data from healthy controls, mild cognitive impairment and Alzheimer’s disease patients with European ancestry originally enrolled in ADNI-GO/2 (n=678). (a) Manhattan plot of −log10 P-values (gray and black) along with quantile–quantile plot of observed versus expected P-values (4 678 609 SNPs were included in the genome-wide interaction study). The green line indicates the genome-wide significance level (P=5 × 10−8). The red dot represents the meta-analysis P-value for rs73069071 using data from ADNI-GO/2 and ADNI-1 samples (P=1.1 × 10−8). (b) Regional visualization of the results for the top SNP (rs73069071; the purple dot; P=6.2 × 10−8) on chromosome 12p12.1. Plot was generated using LocusZoom53 (http://csg.sph.umich.edu/locuszoom/). (c) The position of rs73069071 (located at 21,510,304 in hg19/GRCh37; blue vertical line) and its surrounding genes according to Human GENCODE Annotation in BioDalliance Browser (http://www.gencodegenes.org/human_biodalliance.html).54 IAPP gene (Ensembl gene ID: ENSG00000121351) is located on the forward strand of chromosome 12 (21,507,893-21,532,912). SLCO1A2 gene (Ensembl gene ID: ENSG00000084453) is located on the reverse strand of chromosome 12 (21,417,534-21,572,528). SNP, single-nucleotide polymorphism.
Replication study in participants followed up from ADNI-1 and meta-analysis
We performed replication analyses for rs73069071 and rs112821268 using data from 165 participants who were followed up from ADNI-1 (IMPUTE2 info scores=0.97 and 0.66, respectively). A significant interaction effect was observed for rs73069071 in the same direction observed in the ADNI-GO/2 sample (additive: Pone-tailed=0.028; dominant: Pone-tailed=0.024), while rs112821268 showed no significant interaction effect in the ADNI-1 sample (Pone-tailed=0.44). Hence, all further analyses were carried out only for rs73069071 (major-allele=T; minor-allele=C; minor-allele frequency~12%), which maps to an intronic region within the IAPP (islet amyloid polypeptide or amylin) and the SLCO1A2 (solute carrier organic anion transporter family, member 1A2) genes (Figure 1). Meta-analysis of the results from ADNI-GO/2 and ADNI-1 samples revealed a genome-wide significant (P<5 × 10−8) rs73069071-by-Aβ deposition interaction effect (P=1.1 × 10−8).
Post hoc analyses
In the ADNI-GO/2 sample used in the primary genome-wide analysis, there was no significant difference in demographic and clinical measures between rs73069071C carriers and rs73069071TT homozygotes (Table 1). In the absence of the interaction term in the model, no main effect was observed for rs73069071 genotype on performance in ADAS-cog (P=0.33) or diagnosis (P=0.27), while accounting for the effects of age, sex, years of education and cortical Aβ deposition. Moreover, there was no main effect of the SNP on cortical Aβ deposition (P=0.29).
Incorporating the interaction between rs73069071 genotype and cortical Aβ deposition in the model explained an additional 3.2% of the total variance of performance in ADAS-cog (for comparison purposes: the main effect of APOE genotype in the same model explained 5.2% of the total variance). Excluding the influential observations (n=39; as determined by Cook’s distance >4/number of observations) resulted in further improved interaction effect (P=2.8 × 10−10, n=639). To reveal the nature of the interaction effect, we stratified the groups by genotype. In the ADNI-GO/2 sample, the estimate for the effect size of cortical Aβ deposition on ADAS-cog score controlling for age, sex and education years in rs73069071TT carriers (β=0.49, s.e.=0.05, n=522) was stronger compared with rs73069071C carriers (β=0.19, s.e.=0.08, n=156). Similar results were observed in the ADNI-1 sample (rs73069071TT: β=0.37, s.e.=0.07, n=131; rs73069071C: β=0.21, s.e.=0.13, n=34; after excluding influential observations: Pone-tailed=0.012, n=155). To help the reader visualize the interaction effect, bivariate correlations between cortical Aβ deposition and ADAS-cog score stratified by rs73069071 genotype for both samples are illustrated in Figure 2.

Bivariate correlations between cortical Aβ deposition (as measured by [18F]Florbetapir-PET) and Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog) stratified by rs73069071 genotype in: (a) participants originally enrolled in ADNI-GO/2 (n=678); and (b) participants followed up from ADNI-1 (n=165). Rs73069071CC and rs73069071TC carriers are grouped together for the purpose of visualization.
Rs73069071-by-cortical Aβ deposition interaction was significantly associated with diagnosis (P=2.7 × 10−5) and memory test scores (RAVLT immediate recall: P=8.1 × 10−4, RAVLT learning: P=0.0023 and RAVLT percent forgetting: P=0.0017) in the ADNI-GO/2 sample, all consistent in direction with findings on ADAS-cog. Stratifying the participants based on APOE genotype revealed that the interaction effect was independent of APOE ɛ4 carrier status (ɛ4 carriers: B=−10.3, s.e.=3.8, P=5.5 × 10−4, n=305; ɛ4 non-carriers: B=−14.4, s.e.=3.0, P=3.8 × 10−4, n=373). After stratifying the participants based on diagnostic groups, significant rs73069071-by-cortical Aβ deposition interaction effects in relation to ADAS-cog score were observed within subgroups of AD (B=−10.4, s.e.=4.7, P=0.025, n=107), late MCI (B=−4.4, s.e.=2.1, P=0.03, n=120) and early MCI (B=−4.1, s.e.=2.1, P=0.046, n=254) participants. However, this effect was not evident among the healthy controls (B=−3.0, s.e.=2.0, P=0.14, n=197). The interaction effect remained significant after co-varying for the effect of medication status (medicated with cholinesterase inhibitors and/or memantine vs non-medicated) in AD (92% medicated; P=0.018) and late MCI (39% medicated; P =0.039) patients, and demonstrated trend-level significance in early MCI participants (18% medicated; P=0.06).
In line with our primary finding, exploratory analysis demonstrated a significant rs73069071-by-CSF Aβ1–42 level interaction effect on ADAS-cog score (P=7.2 × 10−4). As expected, the directionality of this effect was opposite to that of the Florbetapir Aβ interaction effect. In addition, we observed a trend-level positive association between rs73069071 C-allele dosage and CSF Aβ1–42 level while controlling for cortical Aβ deposition as well as age, sex and education (P=0.06, n=623). However, this effect was not significant in the absence of cortical Aβ deposition in the model (P=0.49).
Whole-brain cortical thickness analysis
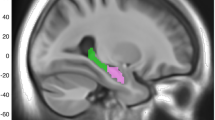
Voxel-based cortical thickness analysis was performed on images from 770 ADNI participants with European ancestry to spatially localize the rs73069071-by-cortical Aβ deposition interaction effect on cortical atrophy. Minor-allele homozygotes were grouped with the heterozygotes to increase the confidence in the results. Consistent with our primary findings on cognitive impairment, we found a significant rs73069071-by-cortical Aβ deposition interaction effect on the cortical thickness of a cluster within the left ventromedial temporal lobe encompassing parahippocampal gyrus, superior and middle temporal gyri, temporal fusiform cortex, temporal pole and amygdala (familywise error corrected P=0.013, cluster volume=12 560 mm3, max X=−23, Y=1, Z=−36, t=4.1). Post hoc analyses showed significant additive (P=6.9 × 10−4) and dominant (P=1.2 × 10−3) rs73069071-by-cortical Aβ deposition interaction effects on the mean cortical thickness of this cluster, while controlling for the effects of age, sex, handedness and education years (Figure 3).

(a) Results for whole-brain voxel-based analysis of rs73069071-by-cortical Aβ deposition interaction effect on cortical thickness depicted in the MNI space (βcortical Aβ deposition in rs73069071 C-allele carriers>βcortical Aβ deposition in rs73069071TT carriers, cluster-wise correction with t>2.3, familywise error corrected P<0.05). We also observed a similar trend in a cluster located in the right ventromedial temporal lobe (familywise error corrected P=0.14, not shown). (b) Bivariate correlations between cortical Aβ deposition and mean cortical thickness of the significant cluster shown in Figure 3a in rs73069071TT (n=598) and rs73069071C (n=172) carriers. Rs73069071CC and rs73069071TC carriers are grouped together for the purpose of visualization.
Longitudinal analyses
Available longitudinal cognitive (ADAS-cog measurements every 6 months up to month 42) and structural MRI (cortical thickness and subcortical volume measurements at baseline and months 3, 6 and 12 from the FreeSurfer v5.1 longitudinal pipeline28) data from ADNI-GO/2 participants were used for longitudinal analyses. Analyzing 2296 observations from 679 participants, we observed significant three-way rs73069071-by-Aβ-by-month of follow-up visit interaction effect on ADAS-cog (Pone-tailed=0.026 [Aβ-by-month of follow-up visit interaction effect in: rs73069071TT: β=0.11, s.e.=0.01, P=4.4 × 10−16; in rs73069071C: β=0.08, s.e.=0.03, P=0.02]) consistent with the results from cross-sectional analysis. We then performed a similar longitudinal analysis on volumes/cortical thickness estimates that passed temporal lobe quality control from the six temporal lobe regions that showed a significant interaction effect in the cross-sectional voxel-based cortical thickness analysis (961 observations from 260 participants; nHealthy controls=87, nearly MCI=107, nlate MCI=51, nAD=15). Consistent with the rs73069071-by-Aβ deposition interaction effect at baseline, we found a significant three-way rs73069071-by-baseline Aβ-by-month of follow-up visit interaction effect on two of these regions of interest (left middle temporal cortical thickness: Pone-tailed=0.019 (Aβ-by-month of follow-up visit interaction effect in: rs73069071TT: β=−0.04, s.e.=0.01, P=0.0004; in rs73069071C: β=0.01, s.e.=0.02, P=0.63) and left amygdala volume: Pone-tailed=0.003 (Aβ-by-month of follow-up visit interaction effect in: rs73069071TT: β=−0.02, s.e.=0.01, P=0.02; in rs73069071C: β=0.02, s.e.=0.01, P=0.09)), while the interaction effect was not significant on parahippocampal gyrus (Pone-tailed=0.31), temporal pole (Pone-tailed=0.49), superior temporal gyrus (Pone-tailed=0.12) and fusiform (Pone-tailed=0.44) cortical thickness measurements.
ROS/MAP postmortem sample
Imputation quality for rs73069071 was high (R2 value=0.96). We did not observe a significant rs73069071-by-Aβ deposition interaction effect (Pone-tailed=0.44) or rs73069071 main effect (P=0.14) on global cognitive function in the ROS/MAP data in the overall sample (n=782). However, we found a significant interaction effect among the AD participants (n=243, Pone-tailed=0.005) consistent in direction with our findings in the ADNI data (Figure 4a). No significant marginal effect was observed for rs73069071 on global cognitive function in AD patients in the absence of interaction term in the model (P=0.62). The interaction effect remained significant after co-varying for medication status in AD patients (32% medicated; P=0.01). However, it was not significant in the MCI patients and healthy individuals. Excluding the influential observations resulted in further improved interaction effects in AD patients (Pone-tailed=7 × 10−4, n=232). Exploratory analysis within the AD participants revealed significant rs73069071-by-diffuse-Aβ deposition interaction effect on cognitive performance (Pone-tailed=0.04; after excluding influential observations: Pone-tailed=5 × 10−5, n=232). However, no interaction effect was evident between rs73069071 genotype and neuritic-Aβ plaques (Pone-tailed=0.2; after excluding influential observations: Pone-tailed=0.07, n=231; Figure 4b). Further analyses revealed significant rs73069071-by-diffuse-Aβ deposition interaction effect on neuritic-Aβ deposition (P=4 × 10−4; after excluding influential observations: P=3.5 × 10−7, n=235). The estimated effect size of diffuse Aβ deposition on neuritic Aβ burden controlling for the effects of age, sex and education was stronger in rs73069071TT participants (β=0.44, s.e.=0.07, n=173) in comparison to rs73069071C carriers (β=0.21, s.e.=0.09, n=70; Figure 4c).

Bivariate correlations in patients with Alzheimer’s disease from ROS/MAP (n=243) stratified by rs73069071 genotype between: (a) brain Aβ deposition (quantitative composite score computed by averaging the percent areas occupied by Aβ across eight brain regions, as measured by immunohistochemistry; Correlations among regional percent areas occupied by Aβ across these brain regions are illustrated in Supplementary Figure 1) and global cognitive function; (b) diffuse (top) and neuritic (bottom) brain Aβ deposition (quantitative composite scores computed by averaging standardized regional densities across 5 brain regions) and global cognitive function; and (c) diffuse and neuritic brain Aβ deposition. Rs73069071CC and rs73069071TC carriers are grouped together for the purpose of visualization.
As ROS/MAP participants were significantly older (mean age=87.7 years) than ADNI-GO/2 participants (mean age=72.5 years), we also performed sub-analyses including only younger healthy, MCI and AD ROS/MAP participants and observed significant rs73069071-by-Aβ deposition interaction effects in participants aged 85 and younger (n=256, Pone-tailed=0.03) (Supplementary Table 1).
Discussion
We identified a genetic variant within the IAPP/SLCO1A2 genes (rs73069071) that modified the effect of cortical Aβ deposition (as measured by [18F]Florbetapir-PET) on AD-related cognitive impairment and temporal lobe atrophy in the ADNI-GO/2 and ADNI-1 samples both cross-sectionally and longitudinally. With greater rs73069071 minor-allele (C-allele) dosage, participants showed weaker associations between brain Aβ deposition and cognitive impairment and temporal lobe atrophy. ROS/MAP postmortem data provided further evidence for the effect of rs73069071 genotype on the relationship between brain Aβ deposition (as measured by immunohistochemistry) and cognitive dysfunction among AD patients. Exploratory analyses suggested that the observed SNP-by-Aβ deposition interaction effect was specific to diffuse-Aβ deposition, rather than neuritic-Aβ plaques. Moreover, AD participants with greater rs73069071 C-allele dosage demonstrated weaker association between diffuse and neuritic Aβ deposition.
Meta-analysis of cross-sectional results in ADNI-1 and ADNI-GO/2 revealed a genome-wide significant IAPP/SLCO1A2 variant-by-in vivo measured cortical Aβ deposition interaction effect on cognitive impairment (ADAS-cog). The interaction effect was evident in all diagnostic subgroups in ADNI-GO/2, except for the healthy participants (that is, AD, late MCI and early MCI; with the largest effect in AD). However, in the ROS/MAP postmortem sample, the rs73069071-by-Aβ deposition interaction effect was present only in AD patients, and was not evident in the whole sample and in the MCI subgroup. This discrepancy between in vivo and postmortem studies may be explained by the differences in the Aβ burden measurements (in vivo PET imaging vs postmortem immunohistochemistry) and/or the correlation between Aβ and cognitive measures (ROS/MAP: r=0.29; ADNI-GO/2: r=0.43). In addition, as also mentioned above, participants in the ROS/MAP sample were significantly older than the participants in the ADNI-GO/2 study. Therefore, the healthy and MCI participants in the ROS/MAP study may represent cognitively healthier sub-populations and might have been more resistant to the effects of Aβ deposition than their counterparts in the ADNI-GO/2 sample. There is also evidence demonstrating that the association between AD pathology and severity of dementia is attenuated in the oldest old, which is suggestive of other underlying neuropathological processes for cognitive dysfunction in this population.29, 30 The fact that we were able to replicate our results in less old ROS/MAP participants further supports this assumption.
Using whole-brain voxel-based cortical thickness analysis, we found a significant IAPP/SLCO1A2 variant-by-cortical Aβ deposition interaction effect on atrophy in AD-susceptible temporal lobe regions (consistent with the primary effect on cognitive impairment). This suggests that IAPP/SLCO1A2 variant may be modifying the impact of Aβ deposition on AD-related neurodegeneration. Critically, we also observed consistent effects longitudinally on both cognitive impairment and atrophy rate in temporal lobe structures. Taken together, these suggest that IAPP/SLCO1A2 variants may have prognostic value predicting brain atrophy and cognitive decline based on cortical Aβ deposition.
Our exploratory analysis in AD participants suggested that the rs73069071 genotype-by-Aβ deposition interaction effect on cognitive dysfunction was primarily driven by the interaction between rs73069071 genotype and diffuse plaque burden. Additionally, we observed stronger association between diffuse and neuritic plaques in rs73069071TT carriers in comparison to rs73069071C carriers. Neuritic and diffuse plaque burden are correlated with one another and both are associated with cognitive impairment and dementia symptoms.31, 32 However, neuritic Aβ deposition is considered to be more closely associated with AD-related neuronal injury,33 while diffuse plaques tend to be less pathogenic34 and occur more frequently in people without dementia.35 Altogether, these data suggest that the rs73069071 C-allele decreases the association between diffuse and neuritic plaque burden, which in turn leads to decreased association between diffuse plaque burden and cognitive impairment. We also observed a trend towards higher CSF Aβ1-42 levels with rs73069071 C-allele dosage in the ADNI-GO/2 data. Therefore, it is also possible that the rs73069071 C-allele mitigates the impairment in Aβ clearance from the brain, which has been shown to be a major culprit in late-onset AD.36
Rs73069071 maps to an intronic region within the IAPP and SLCO1A2 genes on chromosome 12p12.1. No genome-wide significant expression quantitative trait locus or functional variant has been identified in high linkage disequilibrium at this locus (R2>0.4; as per HaploReg v4.1 ([http://compbio.mit.edu/HaploReg)) and therefore definitive molecular consequences of this variant remain to be determined. SLCO1A2 encodes a sodium-independent transporter, which is best known for the cellular uptake of organic anions such as bile acids in the liver. Although SLCO1A2 is highly expressed in the brain,37 we found no evidence supporting its role in AD, or the effect of rs73069071 on SLCO1A2 gene expression levels in different brain regions (n=134).37, 38 However, an IAPP gene product, amylin, has recently been implicated in AD pathophysiology.
The IAPP gene encodes a pancreatic peptide hormone named amylin which is most known for its role in glycemic control. Recent literature has demonstrated that plasma amylin levels are lower in AD and MCI individuals in comparison to healthy controls39, 40 and show positive association with cognitive performance.41 Although it is possible that amylin affects AD-related cognitive impairment through its role in the peripheral metabolic network, recent studies have suggested more direct central effects for amylin.42 Amylin crosses the blood-brain barrier and a recent study has reported amylin deposition in the form of amyloid and also occasional co-depositions of amylin and Aβ in AD brain tissue.43 Although further validation is required, Jackson et al. have proposed that amylin could be considered as the ‘second amyloid’ in AD.43 Amylin oligomerization and deposition in the pancreas of type-2 diabetes patients, which is the result of its chronic hypersecretion, induces apoptotic pancreatic β-cell death.44 Intriguingly, direct neurotoxicity of amyloidogenic forms of amylin (such as the human amylin) have also been documented in embryonic rat hippocampal cultures,45 and in vivo in rats overexpressing human amylin.46
In addition to the direct pathophysiological role of amylin in the brain (neurotoxicity in the form of oligomers and amyloid fibrils), it seems that it is also indirectly involved via interactions with Aβ.40, 47 It is shown that actions of both amylin and Aβ in the brain depend on the amylin receptors48 and that amylin receptor antagonists block both amylin- and Aβ-induced toxicity.49 Moreover, amylin and Aβ are both degraded by the same protease (insulin degrading enzyme).50 In line with these, some recent studies have provided evidence on the beneficial effects of chronic injection of amylin51 and its non-amyloidogenic analog (pramlintide)39, 51 in mice models of AD. Amylin/pramlintide-treated mice demonstrated superior learning and memory performance,39, 51 lower Aβ deposition in the brain,51 increased Aβ in CSF,51 and decreased synapse loss and oxidative stress markers in the hippocampus.39 When taken together, it seems that amylin plays a complex role in the brain and it is possible that while its amyloid form may contribute to AD progression, its non-amyloid form may provide a protective effect.
Our findings must be interpreted in light of the limitations of this study. Despite using the data from relatively large databases with the amyloid burden data, given the frequency of the rs73069071 minor allele (~12%), our sample sizes were relatively small in terms of genetic studies and our findings should be replicated in future studies with larger samples. However, the convergence of the evidence from different modalities and the replication in different samples strengthens the confidence in our findings. The molecular mechanism(s) through which rs73069071 could affect AD pathophysiology is yet to be studied. It is also possible that the effect of rs73069071 on AD is through its effect on genes other than IAPP and SLCO1A2. Assuming that the effect of rs73069071 is on amylin production, assessing the effect of rs73069071 on circulating amylin levels and also simultaneous quantification of Aβ and amylin depositions in characterized AD neuropathology samples such as ROS/MAP or in vivo in humans using PET imaging can shed light on the inter-relationships between Aβ and amylin in determining the risk and progression of AD. As Aβ-PET radiotracers are designed to bind to the β-sheet structure of amyloid depositions, they can also bind to other amyloid aggregates.52 However, the interaction that we observed between the amylin gene polymorphism and Aβ burden in the ADNI sample is unlikely to be driven by a bias of measuring both Aβ and amylin amyloids using [18F]Florbetapir, as [18F]Florbetapir displays high binding affinity (Ki=6.7 nM) for Aβ, but low affinity (Ki=501 nM) for amylin aggregates.52 Furthermore, the interaction effect was also present in the ROS/MAP neuropathology sample where the Aβ burden is quantified by an unbiased immunohistochemistry staining technique. On the other hand, derivatives of Aβ imaging compounds such as [125I]IPBF that show high binding affinity for amylin aggregates52 could potentially be used to measure amylin deposition quantitatively in the brain and contribute to our understanding of late-onset AD pathophysiology.
Overall, our findings provide insight into the complexity of the relationship between Aβ burden and AD-related cognitive impairment. Although not yet functionally validated, our findings support the growing literature on the role of amylin in AD pathophysiology. In addition, pramlintide, which is approved for treatment of diabetes, has been proposed in the treatment of AD.40 Hence, our results could also inform potential clinical trials of pramlintide or similar drugs for AD.
References
Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013; 12: 207–216.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
Musiek ES, Holtzman DM . Three dimensions of the amyloid hypothesis: time, space and'wingmen'. Nat Neurosci 2015; 800–806.
Nelson PT, Braak H, Markesbery WR . Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 2009; 68: 1–14.
Vos SJ, Verhey F, Frolich L, Kornhuber J, Wiltfang J, Maier W et al. Prevalence and prognosis of Alzheimer's disease at the mild cognitive impairment stage. Brain 2015; 138: 1327–1338.
Ma Y, Zhang S, Li J, Zheng DM, Guo Y, Feng J et al. Predictive accuracy of amyloid imaging for progression from mild cognitive impairment to Alzheimer disease with different lengths of follow-up: a meta-analysis. [Corrected]. Medicine (Baltimore) 2014; 93: e150.
Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kövari E, Perl DP et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003; 60: 1495–1500.
Guillozet AL, Weintraub S, Mash DC, Mesulam MM . Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 2003; 60: 729–736.
Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE . Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 2004; 61: 378–384.
Toledo JB, Bjerke M, Da X, Landau SM, Foster NL, Jagust W et al. Nonlinear association between cerebrospinal fluid and Florbetapir F-18 beta-amyloid measures across the spectrum of Alzheimer Disease. JAMA Neurol 2015; 72: 571–581.
Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA et al. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA 2011; 305: 275–283.
Weiner MW, Aisen PS, Jack CR, Jagust WJ, Trojanowski JQ, Shaw L et al. The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement 2010; 6: e207.
Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS . Overview and findings from the religious orders study. Curr Alzheimer Res 2012; 9: 628–645.
Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS . Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res 2012; 9: 646–663.
Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ et al. Amyloid-β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013; 54: 70–77.
Benge JF, Balsis S, Geraci L, Massman PJ, Doody RS . How well do the ADAS-cog and its subscales measure cognitive dysfunction in Alzheimer’s disease? Dement Geriatr Cogn Disord 2009; 28: 63–69.
Estevez-Gonzalez A, Kulisevsky J, Boltes A, Otermin P, Garcia-Sanchez C . Rey verbal learning test is a useful tool for differential diagnosis in the preclinical phase of Alzheimer's disease: comparison with mild cognitive impairment and normal aging. Int J Geriatr Psychiatry 2003; 18: 1021–1028.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009; 65: 403–413.
Yu L, Boyle PA, Leurgans S, Schneider JA, Kryscio RJ, Wilson RS et al. Effect of common neuropathologies on progression of late life cognitive impairment. Neurobiol Aging 2015; 36: 2225–2231.
Hohman TJ, Chibnik L, Bush WS, Jefferson AL, De Jaeger PL, Thornton-Wells TA et al. GSK3β Interactions with Amyloid Genes: An Autopsy Verification and Extension. Neurotox Res 2015; 1–7.
Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA . Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 2012; 72: 599–609.
De Jager PL, Shulman JM, Chibnik LB, Keenan BT, Raj T, Wilson RS et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging 2012; 33: 1017.e1–1017.e15.
Hamshere ML, Walters JT, Smith R, Richards AL, Green E, Grozeva D et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol Psychiatry 2013; 18: 708–712.
Lencz T, Guha S, Liu C, Rosenfeld J, Mukherjee S, DeRosse P et al. Genome-wide association study implicates NDST3 in schizophrenia and bipolar disorder. Nat Commun 2013; 4: 2739.
Nyholt DR, Low SK, Anderson CA, Painter JN, Uno S, Morris AP et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat Genet 2012; 44: 1355–1359.
Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 2013; 340: 1467–1471.
Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK et al. Genetic overlap between Alzheimer's disease and Parkinson's disease at the MAPT locus. Mol Psychiatry 2015; 20: 1588–1595.
Reuter M, Schmansky NJ, Rosas HD, Fischl B . Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 2012; 61: 1402–1418.
James BD, Bennett DA, Boyle PA, Leurgans S, Schneider JA . Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA 2012; 307: 1798–1800.
Haroutunian V, Schnaider-Beeri M, Schmeidler J, Wysocki M, Purohit DP, Perl DP et al. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch Neurol 2008; 65: 1211–1217.
Bennett DA, Wilson RS, Schneider JA, Evans DA, Mendes de Leon CF, Arnold SE et al. Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology 2003; 60: 1909–1915.
Monsell SE, Mock C, Roe CM, Ghoshal N, Morris JC, Cairns NJ et al. Comparison of symptomatic and asymptomatic persons with Alzheimer disease neuropathology. Neurology 2013; 80: 2121–2129.
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012; 8: 1–13.
Masliah E, Terry RD, Mallory M, Alford M, Hansen LA . Diffuse plaques do not accentuate synapse loss in Alzheimer's disease. Am J Pathol 1990; 137: 1293–1297.
Thal DR, Griffin WS, Braak H . Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J Cell Mol Med 2008; 12: 1848–1862.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 2010; 330: 1774.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S et al. The genotype-tissue expression (GTEx) project. Nat Genet 2013; 45: 580–585.
Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014; 17: 1418–1428.
Adler BL, Yarchoan M, Hwang HM, Louneva N, Blair JA, Palm R et al. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer's disease pathogenesis and cognition. Neurobiol Aging 2014; 35: 793–801.
Qiu WQ, Zhu H . Amylin and its analogs: a friend or foe for the treatment of Alzheimer's disease? Front Aging Neurosci 2014; 6: 186.
Qiu WQ, Au R, Zhu H, Wallack M, Liebson E, Li H et al. Positive association between plasma amylin and cognition in a homebound elderly population. J Alzheimers Dis 2014; 42: 555–563.
Lutz TA, Meyer U . Amylin at the interface between metabolic and neurodegenerative disorders. Front Neurosci 2015; 9: 216.
Jackson K, Barisone GA, Diaz E, Lw Jin, DeCarli C, Despa F . Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann Neurol 2013; 74: 517–526.
Epstein FH, Höppener JWM, Ahrén B, Lips CJM . Islet amyloid and type 2 diabetes mellitus. N Engl J Med 2000; 343: 411–419.
May PC, Boggs LN, Fuson KS . Neurotoxicity of human amylin in rat primary hippocampal cultures: similarity to Alzheimer's disease amyloid-β neurotoxicity. J Neurochem 1993; 61: 2330–2333.
Srodulski S, Sharma S, Bachstetter AB, Brelsfoard JM, Pascual C, Xie XS et al. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener 2014; 9: 30.
Fu W, Patel A, Jhamandas JH . Amylin receptor: a common pathophysiological target in Alzheimer's disease and diabetes mellitus. Front Aging Neurosci 2013; 5: 42.
Jhamandas JH, Li Z, Westaway D, Yang J, Jassar S, MacTavish D . Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. Am J Pathol 2011; 178: 140–149.
Jhamandas JH, MacTavish D . Antagonist of the amylin receptor blocks β-amyloid toxicity in rat cholinergic basal forebrain neurons. J Neurosci 2004; 24: 5579–5584.
Shen Y, Joachimiak A, Rosner MR, Tang W-J . Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006; 443: 870–874.
Zhu H, Wang X, Wallack M, Li H, Carreras I, Dedeoglu A et al. Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer’s disease. Mol Psychiatry 2015; 20: 252–262.
Yoshimura M, Ono M, Watanabe H, Kimura H, Saji H . Feasibility of amylin imaging in pancreatic islets with beta-amyloid imaging probes. Sci Rep 2014; 4: 6155.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–2337.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012; 22: 1760–1774.
Acknowledgements
TR is funded by the Isabel Johnson Biomedical Postdoctoral Award, Alzheimer Society of Canada Research Program. AN is funded by the Centre for Addiction and Mental Health and Canadian Institutes of Health Research fellowship awards. DF is supported by a Vanier Canada Graduate Scholarship (CIHR) as well as the Peterborough K.M. Hunter Graduate Scholarship. ANV is funded by the Canadian Institutes of Health Research, Ontario Mental Health Foundation, Brain and Behavior Research Foundation and the National Institute of Mental Health (R01MH099167 and R01MH102324). ROS/MAP is funded by NIA grants P30AG10161, R01AG17917, RF1AG15819.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Alzheimer’s Disease Neuroimaging Initiative Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided the data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Roostaei, T., Nazeri, A., Felsky, D. et al. Genome-wide interaction study of brain beta-amyloid burden and cognitive impairment in Alzheimer’s disease. Mol Psychiatry 22, 287–295 (2017). https://doi.org/10.1038/mp.2016.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.35
This article is cited by
-
Protein Expression of Amino Acid Transporters Is Altered in Isolated Cerebral Microvessels of 5xFAD Mouse Model of Alzheimer’s Disease
Molecular Neurobiology (2023)
-
Aβ efflux impairment and inflammation linked to cerebrovascular accumulation of amyloid-forming amylin secreted from pancreas
Communications Biology (2023)
-
Association of a SNP in the IAPP gene and hyperglycemia on β-cell dysfunction in type 2 diabetes: the Toon Genome Study
Diabetology International (2022)
-
Influence of COMT polymorphism in cognitive performance on dementia in community-dwelling elderly Mexican (SADEM study)
Metabolic Brain Disease (2021)
-
Common Variant in TREM1 Influencing Brain Amyloid Deposition in Mild Cognitive Impairment and Alzheimer’s Disease
Neurotoxicity Research (2020)
