
Abstract
Promoting adult hippocampal neurogenesis is expected to induce neuroplastic changes that improve mood and alleviate anxiety. However, the underlying mechanisms remain largely unknown and the hypothesis itself is controversial. Here we show that mice lacking Jnk1, or c-Jun N-terminal kinase (JNK) inhibitor-treated mice, display increased neurogenesis in adult hippocampus characterized by enhanced cell proliferation and survival, and increased maturation in the ventral region. Correspondingly, anxiety behaviour is reduced in a battery of tests, except when neurogenesis is prevented by AraC treatment. Using engineered retroviruses, we show that exclusive inhibition of JNK in adult-born granule cells alleviates anxiety and reduces depressive-like behaviour. These data validate the neurogenesis hypothesis of anxiety. Moreover, they establish a causal role for JNK in the hippocampal neurogenic niche and anxiety behaviour, and advocate targeting of JNK as an avenue for novel therapies against affective disorders.
Similar content being viewed by others

Introduction
Anxiety and depression are highly prevalent, complex disorders involving multiple brain regions and feedback loops.1 During 2010 alone, anxiety and depressive disorders together comprise the second largest cause of years lived with disability globally, thereby posing a major economic burden.2 Yet, these conditions are mostly undiagnosed and remain untreated because of lack of effective therapies. Anxiety disorders represent a maladaptive state of a normal response to threat, marked by excessive fear and avoidance in the absence of real danger. The hippocampus, classically associated with memory and learning, also controls emotional behaviour, in particular fear and anxiety responses.3, 4, 5, 6 It responds to glucocorticoid stress by exerting neuronal feedback inhibition of the hypothalamic–pituitary axis endocrine response.7 The hippocampus extends dense ventral connections to the prefrontal cortex (PFC) and the basolateral amygdala and shows selective ventral control of the endocrine stress response.8, 9 Moreover, these highly conserved connections are required for anxiety responses.10, 11 These studies, among others,12 have led to the association of the ventral hippocampus with emotional regulation.
Generation of new neurons in the hippocampus throughout adult life (adult hippocampal neurogenesis) is believed to suppress anxiety behaviour.13, 14, 15 Studies in mice have shown that different classes of antidepressant drugs or electroconvulsive seizures (to mimic therapy) enhance levels of neurogenesis in the dentate gyrus (DG) subregion.16, 17, 18 Reports showing that neurogenesis is required for antidepressant drug action13, 19, 20 have led to the theory that promotion of adult hippocampal neurogenesis may alleviate anxiety and depressive disorders. However, this theory remains controversial as other studies have shown beneficial effects of antidepressants independent of new neuron generation,21, 22, 23 calling into doubt the role of new granule cells in regulating mood.
When functioning normally, neurogenesis is an essential part of an adaptive response that buffers the main stress hormone system, the hypothalamic–pituitary axis, and thereby controls behavioural responses in mice8, 18, 24 and in non-human primates.12, 25 Neurogenesis is impaired upon prolonged exposure to excessive glutamate or stress hormones as occurs with anxiety and depression. Its inhibition following prolonged stress results in maladaptive neuroplasticity and reinforces the disease state.1, 26 It is expected therefore that elucidation of the molecular mechanisms that control adult hippocampal neurogenesis will provide new targets for antidepressant and anxiolytic drug therapy. In particular, it has been rationalized that drugs that act on the ventral hippocampus would be of benefit for treatment of anxiety and depression.18 However the intracellular signalling mechanisms governing hippocampal neurogenesis have not been widely explored.
The c-Jun N-terminal kinases (JNKs) are known for their regulation of apoptotic cell death in a variety of cell types.27 Nevertheless, JNK1 is physiologically active in the developing brain where it controls many aspects of cortical development, neuronal differentiation and maturation,28, 29, 30, 31, 32, 33, 34 although its role in adult neurogenesis has not been addressed. More recently, genetic anomalies on the JNK pathway have been associated with psychiatric disorders, although the mechanism is completely unknown.35, 36, 37, 38, 39, 40 Thus, understanding the function of JNK in adult brain has profound implications for the treatment of mental illness.
Here we show that genetic deletion of Jnk1 or intracerebral ventricular infusion with a JNK inhibitor increases neurogenesis in the hippocampus and produces a low anxiety phenotype. Specific retroviral targeting of inhibitor to the hippocampal neurogenic niche cells is sufficient to produce both anxiolytic and antidepressant responses in mice, thereby establishing a causal link. These findings identify JNK1 as a central repressor of neurogenesis and promoter of anxiety and depressive-like behaviour in the adult hippocampus.
Materials and methods
Plasmid construction
To prepare replication-incompetent Moloney murine leukemia virus retroviral vectors expressing cytosolic and nuclear targeted inhibitors of JNK, GFP-NES-JBD and GFP-NLS-JBD (previously described30) were ligated into the AgeI and DraI sites of CAG-GFP (previously described41) to yield CAG-GFP-NES-JBD and CAG-GFP-NLS-JBD.
Reagents
DJNKI-1 peptide inhibitor of JNK was synthesized by Gene Cust, Custom Services for Research (Laboratoire de Biotechnologie du Luxembourg, Dudelange, Luxemburg). Arabinocyl-cytosine (AraC) was from Sigma (St Louis, MO, USA).
Behavioural tests
Adult (8–12-week-old male mice for wild-type/knockout experiments, and 12–16-week-old for chronically infused or retrovirus expressing mice) male C57BL/6J wild-type (WT) and Jnk1−/− mice were maintained on a 12 h light/dark cycle and supplied with food and water ad libitum. Behavioural tests were performed between 0800 and 1500 h in the order described below with 2-day interval between tests. For all procedures experimenters were blind to the genotype or treatments. The number of animals used in each experiment is indicated in the legends. For experiments using Jnk1−/− mice, the cohort numbers are somewhat lower because of the fact that they are poor breeders. Our recount efficiency for behavioural tests with independent experimenters is within 10% variance.
Elevated plus maze
Unconditioned anxiety-like behaviours were monitored using an elevated plus maze (EPM) with two closed (35 × 5 × 15 cm) and two open (35 × 5 cm) arms made of opaque grey plastic elevated at a height of 50 cm above the floor. Mice were placed in the centre of the maze facing a closed arm and allowed to explore freely for 5 min. Behaviour was recorded using the Ethovision video tracking system (Noldus Information Technology, Wageningen, The Netherlands). Automated tracking was used for the analysis of time spent and number of entries to the open and closed arms. Head dipping, rearing and stretched attend postures (risk assessment behaviour) were scored manually from the same videos. Grooming was scored as previously described.42 Note that the behavioural tests in Figure 1 were carried out in Helsinki, whereas behavioural tests in the remaining figures were carried out in Turku. The EPM apparatus in Helsinki had slightly different arm dimensions (30 × 5 × 15 cm).

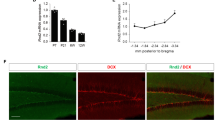
Jnk1−/− mice exhibit low anxiety in an aversive environment and increased adult hippocampal neurogenesis. Wild-type (WT) (n=10) and Jnk1−/− (n=8) mice were tested in the elevated plus maze (EPM), light/dark test and open field test. Several parameters indicative of anxiety behaviour were scored. (a) Jnk1−/− mice spent more time than WT in the open arms of the EPM and less time in the closed arms, indicating reduced anxiety. JNK, c-Jun N-terminal kinase. (b) Representative traces of WT and Jnk1−/− mouse movement on the EPM are shown. (c) Jnk1−/− mice entered the open arms more frequently than WT mice. (d, e) Jnk1−/− mice showed increased head dipping and rearing compared with WT. (f) The average time spent in a light or dark enclosed area is shown. Jnk1−/− mice spent more time in the light area indicating reduced anxiety. In the open field test, Jnk1−/− mice showed (g) reduced latency to enter the centre of the arena and (h) increased time in the centre (shown for six repeated trials per mouse). (i) The overall percent of distance travelled in the centre of the arena is shown. (j) Immobility time in the forced swim test is shown. Jnk1−/− spent significantly less time immobile. (k) The preference of WT (n=12) and Jnk1−/− (n=11) mice for sucrose was tested. Jnk1−/− mice showed a trend towards increased sucrose preference (P=0.05). (l) A representative contour map depicts the dentate gyrus (DG) granular layer region used for Cavalieri volumetric analysis. (m) Quantitative data from Cavalieri estimation of granule layer volume in WT (n=5) and Jnk1−/− (n=5) mice is shown. (n) Representative images of bromodeoxyuridine (BrdU) immunoreactivity in the DG of WT and Jnk1−/− mice 28 days following labelling are shown. (o) Stereological estimate of BrdU-positive cell number in the DG shows a significant increase in Jnk1−/− mice (n=3). (p) The number of doublecortin (DCX)-positive cells in the DG of WT (n=6) and Jnk1−/− (n=7) mice is shown. (q) Representative maximum projection images of DCX staining in the hippocampus of WT and Jnk1−/− illustrates the substantial increase in neurogenic cells in knockout mice. (r) Representative single plane confocal images of BrdU/NeuN labeling in WT and Jnk1−/− mice. Dual-labelled cells are indicated with arrows. (s) The number of BrdU/NeuN double-labelled cells from WT and Jnk1−/− mice is shown (n=3). (t) Representative images of cleaved caspase-3 immunofluorescence staining in the DG of WT and Jnk1−/− mice are shown. Hoechst-33342 (white) and cleaved caspase-3 (green). (u) Quantitated data depicting the number of cleaved caspase-3 cells in WT (n=4) and Jnk1−/− (n=4) mice is shown.
Light/dark
This test was performed in the Helsinki mouse behavioural unit using an Activity Monitor system (for Figure 1) modified with a dark box insert that was opaque to visible light and covered half the area of the open field arena (an opening of 5.5 × 7 cm allowed free movement between the compartments). Animals were placed in the light part of the arena and monitored for 10 min. Time spent in the light and dark areas was calculated. For Figures 2 and 4, a box (30 × 45 × 30 cm), partitioned equally, with opaque black plastic walls (dark) and white walls without roof (light) and an opening (5.5 × 7 cm) between partitions was used. Mice were video monitored for 10 min and analysed manually and also with Ethovision software.

Acute treatment with c-Jun N-terminal kinase (JNK) inhibitor does not alter anxiety related behaviour. (a) The timeline of DJNKI-1 inhibitor or vehicle treatment and behaviour tests are shown along with guide cannula coordinates. DJNKI-1 (100 μM) or vehicle (control) was injected to the ventricles using CMA (Agnthos, Lidingö, Sweden) dialysis at the given coordinates. (b) Hippocampus (HC) and prefrontal cortex (PFC) were isolated from sham operated (control, n=5) and DJNKI-1 (n=5)-treated mice and levels of PSD95 Ser295, PSD95, JNK1 and 2 (JNK1/2) and tubulin were determined by immunoblotting. Representative blots are shown. (c) Quantitative data from multiple immunoblots indicate that PSD95 phosphorylation is significantly reduced in the HC, but not the PFC. NS, not significant. (d–h) Mice treated with vehicle (control, n=5) or 100 μM DJNKI-1 (n=6) were subjected to the elevated plus maze (EPM) test. Behaviour was scored as in Figure 1. (i) Time spent by sham operated (control, n=5) and DJNKI-1-treated mice (n=6) in the light and dark box is plotted.
Open field
Activity in the open field was obtained using the Activity Monitor System (MedAssociates, St Albans, VT, USA). Mice were released in the corner of the open field arena (30 × 30 cm) facing the wall. Horizontal and vertical activities were recorded during 30 min. For the analysis, the arena was divided into a central (18 × 18 cm) and a peripheral zone, and latency to enter the centre, distance travelled in the centre and total distance travelled were scored.
Sucrose preference test
The 8-week-old or 16-week old (for retroviral tranduced mice) singly housed mice were exposed to two drinking tubes in the cage for 3 days. For the test period, bottles containing 12 ml of water or 2% sucrose were placed on opposite sides of the feeding grid. Bottle positions were swapped after weighing at 24 h intervals. Percent sucrose intake was calculated as follows: (Δweightsucrose)/(Δweightsucrose + Δweightwater) × 100.
Forced swim test
At 48 h following the previous test, mice were acclimatized (1 h) and placed in a glass cylinder (18 cm wide × 27 cm deep) filled with 23–25 °C water. Behaviour was recorded using a Samsung SMX-F300BP camera (Suwon, South Korea) for 6 min. Time spent immobile (motionless or only little movements to prevent sinking) was scored from the last 4 min of the recording.
Acute and chronic intracranial infusions
The 8–12-week-old male C57Bl/6J mice were injected intraperitoneally with 0.1 mg kg−1 of buprenorphine hydrochloride (Temgesic, Reckitt Benckiser Pharmaceuticals, Basel, Switzerland), anaesthetized with 4% isoflurane and maintained with 2.5% isoflurane. Viscotears gel (Novartis, Basel, Switzerland) was applied to the eyes to avoid dryness. Local anaesthetic (0.1 ml of 1 mg per 1 μg lidocaine) was subcutaneously injected, before cutting the scalp. For acute intracranial treatments, a CX guide cannula (Agnthos, Lidingö, Sweden) was implanted to the lateral ventricle at the following coordinates, anterior–posterior=−0.6 mm, medial–lateral=1.5 mm and dorsoventral=−2.0 mm, using a stereotaxic alignment instrument (Kopf, Lidingö, Sweden). The guide cannula was fixed to the skull with two screws and dental cement. At 7 days after surgery, 2 μl of DJNKI-1 (100 μM) in saline was injected using a CXMI-5 microinjection cannula (Agnthos) at a rate of 0.25 μl min−1. Behaviour was tested 6 h later. For long-term intracranial infusion of DJNKI-1 or carrier, a small mini-osmotic pump (Alzet Model 2006, Cupertino, CA, USA) was inserted subcutaneously at the back of the neck and attached to a cannula that was implanted into the cerebral ventricle, according to the coordinates above. Mini-osmotic pumps infused DJNKI-1 or TAT (100 μM) at a rate of 0.15 μl h−1. After 6 weeks, mice underwent behavioural testing.
BrdU administration
Bromodeoxyuridine (BrdU; Sigma) was prepared on the day of use by dissolving in saline and injected intraperitoneally 4 times at 2 h intervals to 2-month-old mice (male) at a dose of 75 mg kg−1, as previously described.43 At the designated time, mice were perfused with 4% paraformaldehyde as described below.
Perfusion and tissue processing
Mice were terminally anaesthetized by intraperitoneal injection with pentobarbital (30 mg ml−1 Mebunate, Orion Pharma, Espoo, Finland) and underwent intracardial perfusion with 4% paraformaldehyde as previously described.32 Brains were post-fixed in paraformaldehyde (4%) for 24 h at 4 °C (for all antibodies except doublecortin (DCX) that was post-fixed at room temperature). Sections (40 μm) were cut and collected into phosphate-buffered saline (PBS) using a vibratome (Leica, Wetzlar, Germany), or after cryoprotection in 30% sucrose and freezing in isopentane (Sigma), on a cryostat microtome (CM1950, Leica Biosystems, Nussloch, Germany).
Immunohistochemistry
Free floating sections were immersed in 50% ethanol in PBS followed by permeabilization with Triton X-100 (0.4%) in PBS for 10 min. Endogenous peroxidase was blocked by incubating with H2O2 (3%) in methanol (10%) for 30 min. For BrdU detection, sections were additionally treated with 50% formamide (v/v) in 2 × sodium citrate buffer (0.3 M NaCl and 0.03 M sodium citrate) at 65 °C for 2 h, and then incubated in 2N HCl at 37 °C for 30 min and rinsed in 0.1 M boric acid (pH 8.5) for 10 min as previously described.32 To reduce nonspecific binding, sections were incubated for 1 h with goat serum (10%) in PBS containing Tween-20 (0.2%) or (for green fluorescent protein (GFP) detection) with bovine serum albumin (1%) in PBS containing Triton-X-100 (0.4%). Slices were incubated for 24–72 h at 4 °C with primary antibodies as follows: 1:500 rabbit anti-DCX (Cell Signaling Technologies, Denvars, MA, USA), 1:1000 anti-Prox1 (Millipore, Darmstadt, Germany), 1:100 rabbit anti-Ki67 (#NCL-Ki67, Novocastra, Leica Biosystems, Leica), 1:2000 rabbit anti-GFP (#632460, Cell Signaling Technologies) or 1:200 mouse IgG2a anti-BrdU (#MA3-071, Thermofisher Scientific, Waltham, MA, USA). Biotinylated anti-mouse IgG2a (1:1000) or biotinylated anti-rabbit secondary antibody (1:1000, Vector Laboratories, Burlingame, CA, USA) followed by streptavidin horseradish peroxidase (1:2000) (Invitrogen, Burlingame, CA, USA) were used for signal amplification. For detection, the K3468 kit (Dako, Glostrup, Denmark) was used according to the manufacturer’s instructions.
Immunofluorescence staining
Floating sections were rinsed 3 × 5 min in PBS and incubated for 1 h at room temperature in PBS containing 1% bovine serum albumin and 0.1% Triton-X-100, followed by 72 h of incubation with primary antibody. For BrdU, the formamide treatment described above was also applied. The following primary antibody dilutions were used: 1:1000 mouse anti-BrdU (#AB6326, Abcam, Cambridge, UK), 1:500 mouse anti-NeuN (#MAB377, Millipore), 1:300 rabbit anti-cleaved caspase-3 (#2305-PC, Trevigen,Gaithersburg, MD, USA), 1:2000 anti-GFP (#A6455, Thermofisher, Life Technologies, Carlsbad, CA, USA) and 1:500 goat polyclonal anti-DCX (#8066, Santa Cruz, Dallas, TX, USA). Detection was with Alexa dye-conjugated antibodies (Molecular Probes, Eugene, OR, USA, Thermofisher, Waltham, MA, USA) at a concentration of 1:500. Nuclei were visualized with Hoechst-33342 (1:1000). Tiled images were acquired with a LSM-780 confocal microscope (Carl Zeiss, Oberkochen, Germany) using 10 × , 20 × and 40 × objectives. Images shown are typically from maximum projections of 5 × z-planes.
Stereological analysis
An unbiased estimate of DCX/Ki67/BrdU/GFP or cleaved caspase-3-positive cell number in the subgranular zone of the DG was acquired using every fifth section throughout the rostral–caudal extent of the hippocampus. Exhaustive counting was used for BrdU and cleaved caspase-3, whereas the optical fractionator was used for DCX and Ki67. For fractionator analysis, a 100 × objective, 50 × 50 μm frame size and 220 × 130 μm random sampling grid and 2 μm guard zone were used with the StereoInvestigator (MBF Bioscience, Williston, VT, USA) and Olympus B53 bright-field microscope (Tokyo, Japan). The coefficient of error was <0.1 throughout. For cleaved caspase-3, counting was done from whole right DG using a Leica DMRE fluorescence microscope. Dorsal sections of the DG were defined as sections with coordinates: anterior–posterior=−1.06 mm to –2.80 mm from Bregma and ventral-posterior=−2.80 mm to −3.80 mm from Bregma.
Dendrite analysis
For Sholl analysis, 200 μm coronal sections were cut with a vibratome. Nuclei were visualized with DAPI (4', 6-diamidino-2-phenylindole (Invitrogen). Lucifer yellow dye (Invitrogen) was injected into CA3 pyramidal neurons using a pulled borosilicate glass tube (World Precision Instruments, Sarasota, FL, USA) and a dual μ-iontophoresis current generator (World Precision Instruments, Model 260), with a DC current of 2–6 nA. The three-dimensional images were acquired with a 20 × objective on the Leica TCS SP2 confocal microscope. Sholl analysis was performed using Neurolucida software (Williston, VT, USA) as previously described.33 Dendrite maturation in DCX-positive cells was analysed using the Stereo Investigator (MBF Bioscience) (for DJNKI-1-treated mice) or the LSM-Zeiss-780 (for WT and Jnk1−/− mice). Cells with tertiary dendrites were scored. The maturation index was calculated as follows: (number of DCX-positive cells with tertiary dendrites/number of DCX-positive cells).
Immunoblotting
Brain tissues were snap frozen and stored at −80 °C until denatured using the Denator device (Denator, Uppsala, Sweden). Tissues were homogenized and fractionated on SDS-polyacrylamide gels as previously described.28 For immunoblotting, the following antibody dilutions were used: 1:2000 anti-Ser295-Phospho PSD95 and 1:2000 anti-PSD95 (#3450, Millipore), 1:1000 anti-JNK1/2 (#554285, Pharmingen, San Diego, CA, USA) and 1:10 000 anti-tubulin (#KMX-1, Millipore). Densitometry was performed using the Chemidoc (Bio-Rad, Hercules, CA, USA).
Retroviral production
Viral vectors were produced in HEK-293FT (Thermofisher Scientific) cells as previously described.41 Briefly, 10–12 × 10 cm plates containing 50–70% confluent human embryonic kidney (HEK-293 FT) cells were transfected with retroviral constructs and packaging plasmids pCMV-VSVG and pCMV-GP using Lipofectamine 2000 (Invitrogen). At 5 h after transfection, 10 ml of Dulbecco’s modified Eagle’s medium was replaced with the same volume of fresh, warmed Dulbecco’s modified Eagle’s medium. After 48 h, virus-containing media were collected and briefly centrifuged using an Eppendorf 5804R (Hamburg, Germany) Centrifuge at 2000 r.p.m. for 3 min. The supernatant was filtered using a 0.2 μm Minisart filter (Sartorius, Göttingen, Germany) and centrifuged using a SW32Ti rotor at 19 400 r.p.m. (RCFavg 46 220) in a Beckman Coulter Optima L-90K Ultracentrifuge (Vantaa, Finland) for 2 h at 4 °C. The viral pellet was resuspended in 2 ml sterile PBS and the centrifugation was repeated. The viral pellet was gently resuspended in 100 μl sterile PBS and divided in 10 μl aliquots, snap-frozen in liquid nitrogen and stored at −80 °C until use. Virus titre was determined by seeding HEK-293FT in 24-well plates. When 50–70% confluent, serial dilutions (1:102 to 1:107) of retroviruses were added in 10 μl volumes to wells (in duplicate). Plates were gently rocked before incubation at 37 °C and 5% CO2. After 48 h, wells were rinsed 1 × with sterile PBS, fixed with 4% paraformaldehyde, pH 7.4, for 15–20 min and rinsed 1 × with PBS. Viral titre was obtained by counting the number of cells with detectable GFP using an Olympus IX70 fluorescence microscope. Viral titre, the number of transducing units (TU) per ml, was calculated. Titres ranging from 108 to 1010 TU ml−1 were used for in vivo experiments.
Stereotactic injection of retroviruses
The 8–10-week-old male C57Bl/6J mice were anaesthetized as described above for mini-pump implantations. After 0.1 mg kg−1 of buprenorphine hydrochloride and 4% isoflurane (as above), mice were transferred to a stereotaxic frame (Kopf instruments) and maintained with 2.5% isoflurane and local anaesthetic (0.1 ml of 1 mg per 1 μg lidocaine) was subcutaneously injected, before cutting the scalp. Retroviruses carrying transgenes CAG-GFP, CAG-GFP-NES-JBD and CAG-GFP-NLS-JBD were mixed with polybrene (10 μg ml−1) and injected bilaterally (2 μl per site at a rate of 0.1 μl min−1) at the following coordinates for whole DG injections: anterior–posterior=−2.5 mm from Bregma; medial lateral=1.8 mm; dorsoventral=−2.0 mm and for ventral injections; anterior–posterior=−3.1 mm from Bregma; medial lateral=2.8 mm; dorsoventral=−3.1. The needle was kept in place for 20 min following injection before being withdrawn slowly. The skin was sutured and mice were injected intraperitoneally with saline before being placed on a heating pad until awake. After surgery, animals were single housed and monitored for 4 or 8 weeks as described before behavioural testing. All surgical procedures and behavioural tests were carried out under the approval of the Animal Experiment Board in Finland.
Statistical analysis
P-values were calculated using Student’s two-tailed t-test where two conditions were compared or by one-way analysis of variance (as indicated). In every case, error bars depict s.e.m. P-values are indicated on the graphs. N-values, defined as the number of animals used, are indicated in the legends. Sufficiency of sample size was estimated by post hoc power analysis.
Replication and randomization of animal experiments
Behavioural tests in Jnk1−/− mice were replicated by two independent experimenters on at least two separate cohorts. Stereological data on DCX and BrdU/NeuN in Jnk1−/− mice and following chronic infusion of DJNKI-1 were reproduced by two independent experimenters on at least two separate cohorts. Behavioural data in mice following chronic infusion with DJNKI-1 were reproduced by two independent experimenters on two separate cohorts. Maturity index from DJNKI-1-treated mice was from one animal cohort. Maturity index from WT and Jnk1−/− mice was carried out on one cohort. Sholl analysis of CA3 hippocampal neurons was carried out over a period of ∼2 months as mice became available. Data using retroviruses (Figures 6c–f) came from two separate cohorts carried out by a single experimenter. For some parameters, the phenotype was a trend and became significant when cohorts were merged. Randomization of animals was achieved by assignment of mice to barcoded cages. The log of barcode assignment was only consulted by the experimenter after behavioural testing and/or stereological analysis was completed.
Results
Genetic deletion of Jnk1 reduces anxiety-like behaviour and increases hippocampal neurogenesis in adult mice
Gene anomalies along the JNK pathway have been associated with psychiatric disorders,40 but this finding has not been followed up in mice. We therefore tested whether mice lacking Jnk1 displayed any behavioural irregularities by subjecting them to a battery of tests. This identified a notable low anxiety phenotype. Jnk1−/− mice spent more time in the open arms and less time in the closed arms of an EPM (Figures 1a and b), entered the open arms more frequently (Figure 1c) and displayed increased head dipping and rearing, traits associated with low levels of anxiety (Figures 1d and e). When mice were given the choice between an uncomfortable ‘light’ and customarily preferred, ‘dark’ environment, Jnk1−/− mice showed no preference for the safe dark area. Instead, they spent substantially more time in the illuminated area, indicating a reduced level of anxiety compared with WT mice (Figure 1f). Finally, in the open field test, Jnk1−/− mice exhibited reduced latency to enter the centre of the arena (Figure 1g) and displayed reduced anxiety by spending a greater proportion of time in the centre and travelling a greater distance there (Figures 1h and i). Notably, there was no difference in total distance travelled by Jnk1−/− mice in the EPM, light/dark or open field tests (Supplementary Figures 1a–d). Together, these results demonstrate that mice lacking Jnk1 exhibit a low anxiety phenotype compared with WT mice upon placement in an aversive environment.
We next tested the response of Jnk1−/− mice in the forced swim test, a commonly used test that is interpreted as behavioural despair, with predictive validity for screening of antidepressant drugs.44 Mice lacking Jnk1 showed significantly reduced immobility indicating a low level of depressive-like behaviour (Figure 1j). When tested for sucrose preference, an indicator of anhedonic state in rodents, Jnk1−/− mice showed a trend towards increased sucrose intake (Figure 1k). Together, these results indicate a lower level of depressive-like behaviour in these mice, and that it would be of interest to investigate JNK inhibitors more broadly in depression models.
Anxiety, as well as major depressive disorder, is associated with decreased volume of the hippocampus45 and DG.46 We therefore estimated the volume of the DG granule cell layer in WT and Jnk1−/− mice using the Cavalieri estimator (Figures 1l and m). There was a trend towards increased volume in mice lacking Jnk1. Although this increase remained a trend, the extent of change (6%) precisely matched the proportional decrease observed in the DG volume of patients suffering with major depressive disorder.46 Although it would be of interest to determine whether this has disease relevance, the size effect is small, and a large cohort analysis that is beyond the scope of this study would be required. Moreover, we did not find a correlation between DG volume change and proliferating cells (not shown).
Increased production of new-born granule cells in the DG is associated with reduced anxiety behaviour in mice.14, 15 We therefore tested whether mice lacking Jnk1 displayed altered hippocampal neurogenesis. Dividing cells were labelled in adult mice using the thymidine analogue BrdU, and the number of BrdU-positive cells in the DG was counted 28 days later. This showed an increase in the number of BrdU-positive cells in the DG of Jnk1−/− mice compared with WT mice (Figures 1n and o), suggesting that either cell proliferation and/or survival was enhanced in the absence of Jnk1. In addition, there was a 30% increase in the number of cells committed to a neuronal lineage in the DG of Jnk1-deficient mice revealed by counting the number of cells that stained positive for the neuroblast marker DCX (Figures 1p and q). The regulation of emotional and cognitive function by adult-born neurons dissociates along the dorsal/ventral axis of the DG.18, 47 We therefore examined neuroblast number in dorsal and ventral dentate gyrus. DCX cell number was significantly increased in both regions in mice lacking Jnk1 (Figure 1p). To determine whether there was an increase in newly generated neurons in these mice, we measured the number of BrdU-positive cells that expressed the mature neuronal marker NeuN at 28 days after labelling. This revealed that the total number of adult-born neurons increased in the DG of Jnk1−/− mice (Figures 1r and s).
Promoting the survival of adult-born neurons in the DG is essential if they are to integrate and contribute functionally to the hippocampal circuitry. Therefore, to specifically evaluate survival in the hippocampal niche, we used an antibody that recognized the cleaved form of caspase-3, but not the pro-enzyme, to measure the extent of apoptotic death. As expected,48 cleaved caspase-3-positive cells were mostly observed in the subgranular zone. Stereological examination revealed a significantly reduced number of cleaved caspase-3-positive cells in Jnk1−/− mice. This result (Figures 1t and u), together with the significant increase in BrdU-positive cell number (Figure 1o), provides a strong indication that survival is increased in the absence of Jnk1 in the hippocampal neurogenic niche.
Acute inhibition of JNK1 does not alter anxiety behaviour
Developmental irregularities in the central nervous system of Jnk1−/− mice are well characterized. These include cortical migration and commissure defects,29, 32, 49 structural changes that could in principle influence behaviour. To overcome this limitation with Jnk1 knockout mice, we utilized a peptide inhibitor of JNK to directly inhibit the kinase in adult brain. We started by examining the effect of acute inhibition of JNK on anxiety-like behaviour. Mice underwent intracerebral–ventricular infusion with vehicle or with a D-amino acid retro-inverso inhibitor of JNK tethered to a TAT delivery sequence. This peptide binds to JNK1 with high affinity providing specific inhibition of kinase activity.50 DJNKI-1 (100 μM) was delivered directly to the ventricles 1 week following cannula implantation, and behaviour was tested 6 h after injection (Figure 2a). The inhibitor effectively blocked JNK substrate phosphorylation in the hippocampus, as monitored by phosphorylation of the JNK1 substrate PSD95 on S295, but robust inhibition was not detected in the PFC (Figures 2b and c). Even though DJNKI-1 diminished S-295 phosphorylation of PSD95 in the hippocampus, this acute treatment did not significantly alter behaviour of mice subjected to the EPM or light/dark test (Figures 2d–i). These data suggested that another process such as neurogenesis, which takes place over a period of several weeks, may underlie the low anxiety behaviour in Jnk1−/− mice.
Long-term infusion with a JNK inhibitor (DJNKI-1) increases neurogenesis in the ventral DG and lowers anxiety
We next tested whether chronic treatment with DJNKI-1 altered anxiety-like behaviour in mice exposed to aversive conditions. Mice were fitted with mini-pump implants and infused with DJNKI-1 or vehicle for 6 weeks (Figure 3a). DJNKI-1 effectively reduced JNK activity in the hippocampus but not in the PFC, as assessed from PSD95-S295 phosphorylation (Figures 3b and c). One possible reason for this absence of effect in the PFC is a deficiency of DJNKI-1 binding sites in that region compared with the hippocampus. This would be consistent with low JNK expression in the cortical lobes in general compared with the hippocampal formation.51 When the effect of inhibitor on anxiety responses was tested, DJNKI-1-treated mice exhibited several behavioural traits symptomatic of low anxiety (Figures 3d–g). They entered the open arms of the EPM more frequently and spent more time there than did vehicle-treated mice (Figures 3d–f). Inhibitor-treated mice also showed increased head dipping and fewer stretch-attend postures than control mice. They reared more often and groomed less, activities indicative of low anxiety (Figure 3g).

Longitudinal treatment with DJNKI-1 lowers anxiety-like behaviour and increases adult hippocampal neurogenesis. (a) A schematic of experimental timeline for treatments is shown. Mice were implanted with osmotic mini-pumps filled with vehicle or DJNKI-1 (100 μM). (b) The effect of inhibitor treatment on c-Jun N-terminal kinase (JNK) substrate phosphorylation was tested by immunoblotting for PSD95-S295 phosphorylation. Representative blots are shown. (c) DJNKI-1 significantly inhibited PSD95-S295 phosphorylation in the hippocampus (HC) but not in the prefrontal cortex (PFC) (n=3 respectively). NS, not significant. (d) The effect of long-term treatment with DJNKI-1 (n=17) or vehicle (n=16) on mice subjected to the elevated plus maze (EPM) was scored, as in Figure 1. The number of entries to the open arms (e, f) and time spent in the arms including representative traces are shown. (g) Head dipping, stretch attend (Str. Attend), rearing and grooming behaviour were scored from the same mice while undergoing EPM testing. (h) Representative maximum projection images from the dentate gyrus of control (n=5) and DJNKI-1 (n=5)-treated mice immunostained for doublecortin (DCX). Scale bar, 100 μm. (i) The estimated total number of DCX-positive cells in the dentate gyrus (DG) in vehicle and inhibitor-treated mice is shown. (j) The estimated number of DCX-positive cells in the dorsal and ventral DG is shown. (k) The number of bromodeoxyuridine (BrdU)/NeuN-positive neurons in the entire DG, (l) and in the dorsal and ventral DG from control (n=5) and DJNKI-1 infused (n=5) mice is indicated. (m) A representative confocal section through nuclei of BrdU/NeuN-stained control and DJNKI-1-treated mouse DG is presented. (n) The number of cleaved caspase-3-positive cells in the entire DG (o) and the dorsal and ventral DG is shown for vehicle (n=4) and DJNKI-1-infused (n=4) mice. (p) A representative image of cleaved caspase-3 staining is shown.
In addition to its anxiolytic effect, DJNKI-1 increased the number of DCX-positive neuroblasts in the DG (Figures 3h and i), consistent with the increase observed in Jnk1−/− mice (Figures 1p and q). In contrast to Jnk1−/− mice, however, the increase in neuroblast number was only apparent in the ventral hippocampus (Figure 3j). The proportional increase upon DJNKI-1 treatment was very similar to that observed in Jnk1−/− mice (Figures 1p and q), indicating that JNK1 represses neurogenesis.
There was also an increase in BrdU/NeuN-positive cells in DJNKI-1-treated mice compared with vehicle-treated controls (Figure 3k), indicating that survival and/or maturation of neurons was increased following infusion with JNK inhibitor. When we examined the spatial distribution of these newly generated neurons along the dorsal ventral axis, we found that the increase in BrdU/NeuN-positive cell number was restricted to the ventral DG (Figures 3l and m). Interestingly however, when we examined cell survival in DJNKI-1-treated mice, we observed that DJNKI-1 exerted a more or less uniform neuroprotective effect throughout the dorsal and ventral regions of the DG (Figures 3n–p). Taken together, these data indicate that JNK inhibition increases neurogenesis in the ventral hippocampus and reduces anxiety-like behaviour in response to an aversive environment.
The anxiolytic effect of DJNKI-1 is dependent on adult neurogenesis
To determine whether increased neurogenesis was critical for the low anxiety behaviour, we infused mice with the mitotic inhibitor AraC at 2%, a dose that blocks cell division, without interfering with the survival of nondividing cells52 (Figure 4a). Infusion with AraC prevented the anxiolytic effect of DJNKI-1 in the elevated plus maze (Figures 4b and c). AraC also prevented the DJNKI-1-evoked anxiolytic effect measured by head dips (Figure 4d) and grooming (Figure 4e). Interestingly though, AraC alone reduced scores for risk assessment behaviour (Figure 4f), it did not increase willingness to explore an unprotected (Figures 4b–e) or illuminated (Figures 4g and h) area, key indicators of lower anxiety. Thus, the effect of AraC on stretch attend postures alone cannot be interpreted as anxiolytic. Together, these results suggest that the anxiety-lowering effect of DJNKI-1 is dependent on neurogenesis.

Treatment with the mitotic inhibitor AraC blocks neurogenesis and the anxiolytic effect of DJNKI-1. (a) A schematic of the experimental set-up showing the timeline of bromodeoxyuridine (BrdU) and inhibitor infusions. The 2-month-old mice were implanted with osmotic mini-pumps filled with vehicle (control) (n=6), 100 μM DJNKI-1 (n=7), 2% AraC (n=6) or 100 μM DJNKI-1 and 2% AraC combined (n=7). After 6 weeks of infusion, mice were subjected to behaviour testing. Behaviour in the elevated plus maze was scored according to the (b) number of entries to the open arms, (c) time spent in the arms, (d) the number of head dips, (e) the number of grooming events and (f) the number of stretch-attend postures. NS, not significant. (g, h) The time spent by mice in the light arena and the latency to enter the dark arena is shown. (i, j) The estimated number of doublecortin (DCX)-positive cells in the dorsal and ventral dentate gyrus is show. (k) The estimated total number of DCX-positive cells following DJNKI-1 infusion is shown. DJNKI induced a significant increase in DCX-positive cell number. P-values were calculated by one-way analysis of variance (ANOVA).
To determine whether AraC infusion had efficiently blocked cell proliferation as intended, we analysed the number of cells that were positive for Ki67 that marks active phases of the cell cycle. AraC efficiently ablated cell proliferation throughout the DG, as expected. Interestingly, DJNKI-1 increased cell proliferation throughout the dorsal–ventral axis of the hippocampus (Supplementary Figures 2a–c). As seen earlier (Figure 3j), DCX-positive cells were prominently increased in the ventral region by DJNKI-1 (Figures 4i–k, Supplementary Figure 2d). Taken together, these results indicate that DJNKI-1 increases the proliferating pool of cells equally in the dorsal and ventral dentate gyrus, whereas promotion of neurogenesis is graded along the dorsal–ventral axis, the largest increase being observed in the ventral region.
JNK inhibition promotes maturation of newborn neurons in the adult DG
Dendritic branching is a fundamental feature of maturing neurons that enables propagation of electrochemical signals and neuroplastic changes.53 Given that JNK1 regulates dendrite arborization in the cortex and cerebellum,30, 33, 38 we investigated whether inhibition of JNK promoted dendritic arborization of adult-born neurons in the DG. To do this, the number of DCX-positive cells with third-order dendrites was counted (Figures 5a–f). DJNKI-1 treatment enhanced the maturation index of new neurons in the ventral but not the dorsal hippocampus (Figures 5a–c), whereas in Jnk1−/− mice, the complexity of DCX cell dendrites was increased in both dorsal and ventral hippocampus (Figures 5d–f), consistent with the uniform regulation of neuroblast number along the longitudinal axis in these mice (Figures 1p and q). Newborn granule cells, upon maturation, provide excitatory input to CA3 pyramidal neuron apical (and to a lesser extent basal) dendrites (as well as to interneurons in the hilus), thereby forming the second synapse of the hippocampal trisynaptic circuit.54 As synaptic innervation provides a stimulus that maintains and elaborates dendritic arbors,55 we measured CA3 pyramidal cell dendritic complexity as an indirect measure of innervation. Consistent with the increased number of newborn granule cells in Jnk1−/− mice (Figure 1r), the dendritic field size of CA3 pyramidal neurons was increased (Figures 5g–l). This suggests functional integration of the newborn neurons. Nonetheless, we do not exclude the possibility that JNK1 regulation of CA3 dendrites has an intrinsic component as we and others have previously shown in the cerebellum and cortex.29, 30, 33

Dendrite arborization is increased in the ventral dentate gyrus (DG) of DJNKI-1-treated mice. (a, b) The number of doublecortin (DCX)-positive cells with tertiary dendrites is shown from the dorsal and ventral DG in sham operated (control, n=4) and D-JNKI-1-treated (n=5) mice. JNK, c-Jun N-terminal kinase. The ‘maturation index’ (the number of DCX-positive cells with tertiary dendrites/number of DCX-positive cells) is also plotted. (c) Representative images of DCX staining from the ventral hippocampus are shown. Scale bar, 50 μm. (d) The number of DCX-positive cells with dendrites or (e) the maturation index in wild-type (WT; n=4) and Jnk1−/− (n=4) mice is shown. P-values (a–d) were calculated using Student’s t-test. (f) A representative image of DCX staining in the ventral hippocampus. Scale bar, 20 μm. Sholl analysis performed on slices from WT and Jnk1−/− hippocampal CA3 pyramidal cells depicts the number of intersections, and length of apical (g, h) and basal (i, j) dendrites. (k) Total dendritic field length and (l) the number of nodes per dendrite is shown. The number of cells analysed for WT and Jnk1−/− mice (g–l) are indicated on the figure. For CA3 apical dendrites, 15 cells each from 4 WT and 4 Jnk1−/− mice were analysed. For CA3 basal dendrites, 32 cells each from 3 WT and 3 Jnk1−/− mice were analysed. P-values were calculated with one-way analysis of variance (ANOVA). Fv (values of variance) and Fc (critical F-values) are shown.
Inhibition of JNK in adult-born neurons of the hippocampus using a retroviral delivery elicits an anxiolytic response
The use of the mitotic inhibitor AraC (Figure 4) to determine a causal relationship between adult-born hippocampal neurons and behaviour is not unequivocal as infused inhibitor is not restricted to the DG and AraC is associated with other nonspecific effects in neurons.56 To establish more specifically whether new neurons in the hippocampus contribute to behaviour, we generated retroviral tools that deliver JNK inhibitor to dividing cells in the hilus. Under the control of a CAG promoter, viral transgene expression is restricted to proliferating cells destined to become new neurons (Figure 6).57 We utilized both nuclear and cytosolic-targeted JNK inhibitor sequences (previously characterized),30, 58 as JNK1 is known to phosphorylate nuclear transcription factors and cytoskeletal regulator targets in the cytoplasm.34 Compartment-specific localization of tagged inhibitors was first validated in 293FT cells that had been virally transduced (Figure 6a). Similarly, stereotactic injection of viruses to the DG followed 5 weeks later by analysis of labelled cells showed compartment-specific labelling of cells in the dorsal and ventral hippocampus (Figures 6b and c, insets). Behavioural testing 4 weeks following injection revealed a significant effect of GFP-NLS-JBD on anxiety-like behaviour in the EPM. Inhibition of JNK in the nuclear compartment induced a significant anxiolytic response (Figures 6d and e), whereas GFP-CAG-NES-JBD did not significantly alter EPM behaviour, with the exception of rearing (Figure 6e). These results indicate that JNK activity in the nucleus of adult-born hippocampal neurons serves a critical function in the promotion of anxiety behaviour in mice.

Inhibition of nuclear c-Jun N-terminal kinase (JNK) in adult-born neurons of the hippocampus produces an anxiolytic effect. (a) Representative confocal micrographs of HEK-293-FT cells infected with retroviruses encoding nuclear (CAG-NLS-JBD) and cytosolic (CAG-NES-JBD) targeted JNK inhibitor transgenes or CAG-GFP are shown (green). Hoechst-33342-stained nuclei (blue). Scale bar, 50 μm. (b) A schematic representation depicting the experimental timeline. Mice were injected with high titre (108–1010 TU) retroviruses (encoding CAG-GFP, CAG-NLS-JBD or CAG-NES-JBD) at day 0. After 4 weeks, mice were tested in the elevated plus maze (EPM) or forced swim test (FST). After killing, bilateral expression of viral inserts was validated using green fluorescent protein (GFP) detection. (c) Maximum projection images of coronal sections through the dentate gyrus (DG) from mice expressing CAG-GFP (upper panels), CAG-NLS-JBD (middle panels) or CAG-NES-JBD (lower panels) are shown. Scale bar, 50 μm. Insets show single plane confocal sections through the nucleus. (d, e) The behaviour of retrovirus-infected mice on the EPM is shown, as before. Mice expressing the nuclear JNK inhibitor display a range of low anxiety behaviour compared with control mice expressing CAG-GFP. The number of mice scored was as follows: CAG-GFP; n=13; CAG-NLS-GFP; n=14; GFP-NES-JBD; n=11. (f) The immobility of mice in the FST following 4 weeks of transgene expression is shown. (g) A schematic representation depicting the 8-week experimental timeline. (h) The immobility behaviour of retrovirus-transduced mice in the FST is shown. The number of mice scored for this timeline was as follows: CAG-GFP; n=8; CAG-NLS-JBD; n=10; CAG-NES-JBD; n=7. (i) The sucrose preference of these mice is shown. (j) The total count of GFP-positive cells in the DG of retrovirus-infected mice whose behaviour was described (g–i) is indicated. For all behaviour experiments, only mice showing bilateral GFP expression were included in final scoring. No other exclusions were made.
Retroviral-based inhibition of JNK in adult-born granule cells reduces depressive behaviour
Given that Jnk1−/− mice exhibited a low depressive phenotype (Figure 1j), we next tested the effect of retroviral JNK inhibitors on mice undergoing the forced swim test of behavioural despair. The 4-week expression of GFP-CAG-NLS-JBD or GFP-CAG-NES-JBD did not alter immobility time in this test of behavioural despair (Figure 6f), although there was a slight trend. We therefore tested the impact of 8 weeks of inhibitor expression. Following 8 weeks of expression, the nuclear JNK inhibitor significantly reduced immobility time (Figure 6h), indicating reduced depressive behaviour. Consistent with this, mice expressing the nuclear JNK inhibitor in newly generated neurons only showed a significant increase in sucrose preference with no increase in overall liquid intake (Figure 6i), indicating a lower anhedonic state. Interestingly, expression of the cytosolic JNK inhibitor showed a trend towards lower depressive activity in both tests (Figures 6h and i), possibly indicating a secondary role. To determine the number of adult-born neurons that were targeted using this approach, we carried out a stereological count of GFP-positive cells in the DG of infected mice. These numbers (Figure 6j), illustrate that <200 new neurons per DG was sufficient to elicit a significant change in behaviour. Together, these data indicate that inhibiting of JNK1 activity in the nucleus of just a few hundred newly generated neurons in the hippocampus alleviates anxiety and depressive-type behaviour in mice. This identifies the JNK pathway as a novel target worthy of further exploration for treatment of anxiety and depressive disorders.
Discussion
Here we identify that Jnk1−/− mice display a low anxiety phenotype and increased hippocampal neurogenesis. Intracerebral infusion of adult mice with a specific JNK inhibitor50 also lowers anxiety and enhances neurogenesis and dendrite elaboration, essential adaptive responses to stress. Remarkably, targeted inhibition of JNK in just a few hundred newly born granule cells of the DG using retroviruses is sufficient to lower anxiety and depressive behaviour. Together, these data emphasize the importance of adult-born granule cells in the hippocampus for the control of anxiety and depressive-like behaviour and identify that JNK controls these behaviours centrally from the hippocampal neurogenic niche. We propose therefore that inhibition of JNK1 should be considered as a new avenue for therapeutic targeting of affective state disorders.
Neurogenesis can be broken down into several stages: cell proliferation, differentiation, neuronal maturation and survival.59 We find that all four processes are promoted by genetic deletion of Jnk1 or peptide inhibition. Cell proliferation is increased by 80% and survival by 200% at 6 weeks following JNK1 inhibitor treatment (Figure 3 and Supplementary Figure 2). These changes occur in both the dorsal and ventral hippocampus. Although all neurogenesis stages are expected to contribute to the anxiolytic effect, survival promotion upon JNK inhibition is likely to be critical,59 and is reminiscent of classical JNK action in developing brain where it elicits pro-apoptotic effects that fine-tune cell number.60, 61 This neuroprotective effect of JNK inhibition combined with enhanced progenitor cell proliferation provides a powerful neurogenic stimulus that ameliorates anxiety behaviour.
Functional integration of adult-born neurons within the hippocampal circuitry defined by dendritic innervation is important if these cells are to effect hippocampal function.62, 63 It is significant therefore that we detect enhanced arborization of newly generated granule cells following infusion with inhibitor for 6 weeks. This enlarged dendritic field will maximize the potential for innervation of newborn granule cells by excitatory input from the entorhinal cortex.57 In addition, that we observe enhanced arborization of CA3 pyramidal cell dendrites in Jnk1−/− mice indicates that functional integration is increased upon loss of JNK1 signalling.
Using retroviruses to singly target JNK in newly generated neurons of the hippocampus, we show that inhibition of nuclear JNK lowers anxiety (EPM), behavioural despair (forced swim test) and anhedonia. Thus, inhibition of JNK in a few hundred adult-born granule cells is sufficient to evoke behavioural improvement, even without expansion of the granule cell population. To our knowledge, this is the first time that the adult-born granule cell population has been singly targeted enabling precise definition of its role in anxiolytic and antidepressant outcomes. Previous approaches, thymidine kinase/ganciclovir-induced apoptosis or irradiation, have targeted dividing cells of the niche,15, 64 whereas here JNK inhibitor expression is introduced to the hilar cells using replication-incompetent retroviruses, thereby avoiding the need for frequent ganciclovir injections or more invasive strategies. Retroviruses only infect dividing cells and JNK inhibitor expression, driven by a CAG promoter, is strongly induced once dividing cells become neurons.57, 65, 66 In this way, selective labelling of adult-born neurons is achieved and post-mitotic early-born neurons or astrocytes do not receive inhibitor. Consistent with this, we only observe GFP expression in cells of the granule cell layer that display granule cell morphology (including mossy fibre labelling), and are positive for Prox1, a marker of granule cells67 (Supplementary Figure 1). Thus, it seems improbable that the anxiolytic effect of CAG-NLS-JBD in retroviral labelling experiments emanates from dividing progenitors. We would like to point out however that the overall effect size of JNK inhibition on anxiety behaviour is larger when the proliferating cells of the niche are also targeted, leading to increased numbers of adult-born neurons (Figures 1 and 3). Together, these findings suggest two molecular checkpoints at which JNK regulates affective behaviour from the hippocampal neurogenic niche. First, it acts to repress proliferation and survival of the dividing cell population; second, it acts within the newly generated post-mitotic granule cells to regulate anxiety and depressive behaviour.
The dominant effect of nuclear JNK revealed by this study is consistent with the previously reported neuroprotective action of nuclear, not cytosolic, JNK inhibition.68, 69 This defines a primarily nuclear locus for the antidepressant and anxiolytic action of JNK inhibitors in the hippocampal neurogenic niche. Similarly, the anxiolytic outcome upon infusion with DJNKI-1 is likely to depend on nuclear JNK inhibition, as DJNKI-1 accumulates passively in neuronal nuclei.70 Although we observe decreased phosphorylation of the cytosolic JNK1 target PSD95 upon inhibitor treatment, the less prominent effect on behaviour of cytosolic JNK inhibitors suggests that this is not the principal mechanism whereby JNK regulates anxiety. The importance of nuclear JNK in regulating adult neurogenesis is entirely consistent with the many transcription factor targets of JNK.71 These include several members of the bZIP and bHLH families as well as the glucocorticoid receptor71 that together can lead to complex gene regulation with diverse cellular outcomes. Nuclear JNK most probably acts via transcriptional programs to suppress the different stages of neurogenesis noted here, that is, proliferation, differentiation, survival and maturation, as inhibition of JNK in the cytosol is insufficient to evoke a significant amelioration of behaviour.
The hippocampus is functionally divided along its dorsoventral axis. The ventral (corresponding to anterior in primates) subregion connects to the amygdala and PFC12, 72 and is associated with emotional behaviour and affective disorders,5, 8, 47, 73 whereas the dorsal region (corresponding to posterior in primates) is associated with spatial learning.74, 75 In non-human primates, elevated activity in the lateral anterior hippocampus is associated with anxiety in individuals that display increased hypothalamic–pituitary axis activity,25 consistent with the ventral-specific endocrine regulation. In humans with both global and somatic anxiety, increased connectivity is observed between the anterior hippocampus and the amygdala.76 These studies provide impetus to examine dorsal and ventral hippocampus in rodent studies, where causative associations can be tested. Consistent with the clinical findings, we detect a more prominent increase in the generation and maturation of neurons in ventral hippocampus following inhibitor treatment, even though proliferation and survival increase throughout the dorsoventral axis. One possible explanation for this ventral bias could be an opposing contribution from JNK1 and JNK2/3 isoforms towards later stages of neurogenesis. By homology to LJNKI-1, DJNKI-1 should inhibit all JNK isoforms.77 JNK2/3 isoforms, like JNK1, are physiologically active in adult hippocampus,78 where they are required for stress-induced amnesia,79 a process requiring adult hippocampal neurogenesis in the dorsal hippocampus.80 In support of this proposal, there is a less prominent dorsal–ventral gradient for elevated neurogenesis in Jnk1−/− mice as compared with inhibitor treated. Regardless of differential JNK isoform contributions, the ventral bias following inhibitor treatment indicates that JNK inhibition provides an ideal approach to preferentially promote neurogenesis in the ventral hippocampus, with potential benefit for the treatment of affective disorders.18
The ventral hippocampus has reciprocal connections to the medial PFC.81 Abnormal interaction between the ventral hippocampus and the medial PFC is not only associated with depression, but also with schizophrenia and post-traumatic stress disorder.72 Interestingly therefore, sequence variation in MKK7, an upstream JNK pathway regulator, have been associated with schizophrenia.39 Although it is not clear whether these anomalies result in a gain or loss of JNK function, the hippocampal connection described here may contribute.
Here we report that JNK is a dominant controller of anxiety and depressive behaviour and that inhibition of the JNK pathway is accompanied by increased neurogenesis and relief from depressive and anxiety-like behaviour. The graded plasticity changes upon JNK inhibition, highest in ventral hippocampus, may be disease relevant as neuroimaging studies have revealed increased functional connectivity in the anterior hippocampus and amygdala of patients with anxiety, implicating this region in the pathophysiology of mood disorders.76, 82 Consistent with this proposal, neuronal JNK is activated by stressors that are considered central in anxiety and depressive disorders including corticosterone83 and glutamate, acting via NMDA (N-methyl-D-aspartate) or AMPA receptors.77, 84, 85 Significantly, glutamate and cortisol in humans and corticosterone in rodents impair neurogenesis.1, 86 In addition, JNKs are activated in animal models of depression and regulate glucocorticoid receptor signalling.86, 87, 88 Our data identify a mechanism whereby JNK may play a central role in the signalling events underlying affective disorders and highlight a new benefit of targeting a protein kinase that is widely associated with apoptotic signalling.
References
Gold PW . The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry 2015; 20: 32–47.
Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380: 2163–2196.
Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 1995; 152: 973–981.
MacMillan S, Szeszko PR, Moore GJ, Madden R, Lorch E, Ivey J et al. Increased amygdala: hippocampal volume ratios associated with severity of anxiety in pediatric major depression. J Child Adolesc Psychopharmacol 2003; 13: 65–73.
Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB . Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci USA 2002; 99: 10825–10830.
Kheirbek MA, Drew LJ, Burghardt NS, Costantini DO, Tannenholz L, Ahmari SE et al. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron 2013; 77: 955–968.
Radley JJ, Kabbaj M, Jacobson L, Heydendael W, Yehuda R, Herman JP . Stress risk factors and stress-related pathology: neuroplasticity, epigenetics and endophenotypes. Stress 2011; 14: 481–497.
Bannerman DM, Rawlins JN, McHugh SB, Deacon RM, Yee BK, Bast T et al. Regional dissociations within the hippocampus—memory and anxiety. Neurosci Biobehav Rev 2004; 28: 273–283.
Strange BA, Witter MP, Lein ES, Moser EI . Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 2014; 15: 655–669.
Forster GL, Novick AM, Scholl JL, Watt MJ . The role of the amygdala in anxiety disorders. In: Ferry B (ed). The Amygdala - A Discreet Multitasking Manager. In-tech, Rijeka, Croatia, 2012, pp 61–102.
Padilla-Coreano N, Bolkan SS, Pierce GM, Blackman DR, Hardin WD, Garcia-Garcia AL et al. Direct ventral hippocampal-prefrontal input is required for anxiety-related neural activity and behavior. Neuron 2016; 89: 857–866.
O'Leary OF, Cryan JF . A ventral view on antidepressant action: roles for adult hippocampal neurogenesis along the dorsoventral axis. Trends Pharmacol Sci 2014; 35: 675–687.
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 2009; 62: 479–493.
Revest JM, Dupret D, Koehl M, Funk-Reiter C, Grosjean N, Piazza PV et al. Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol Psychiatry 2009; 14: 959–967.
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA . Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011; 476: 458–461.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS . Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 2000; 20: 9104–9110.
Surget A, Tanti A, Leonardo ED, Laugeray A, Rainer Q, Touma C et al. Antidepressants recruit new neurons to improve stress response regulation. Mol Psychiatry 2011; 16: 1177–1188.
Tanti A, Belzung C . Neurogenesis along the septo-temporal axis of the hippocampus: are depression and the action of antidepressants region-specific? Neuroscience 2013; 252: 234–252.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003; 301: 805–809.
Schloesser RJ, Orvoen S, Jimenez DV, Hardy NF, Maynard KR, Sukumar M et al. Antidepressant-like effects of electroconvulsive seizures require adult neurogenesis in a neuroendocrine model of depression. Brain Stimul 2015; 8: 862–867.
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 2009; 14: 764–773, 739.
Marlatt MW, Lucassen PJ, van Praag H . Comparison of neurogenic effects of fluoxetine, duloxetine and running in mice. Brain Res 2010; 1341: 93–99.
Deng W, Gage FH . The effect of immature adult-born dentate granule cells on hyponeophagial behavior is related to their roles in learning and memory. Front Syst Neurosci 2015; 9: 34.
Fanselow MS, Dong HW . Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010; 65: 7–19v.
Shackman AJ, Fox AS, Oler JA, Shelton SE, Davidson RJ, Kalin NH . Neural mechanisms underlying heterogeneity in the presentation of anxious temperament. Proc Natl Acad Sci USA 2013; 110: 6145–6150.
Musazzi L, Treccani G, Mallei A, Popoli M . The action of antidepressants on the glutamate system: regulation of glutamate release and glutamate receptors. Biol Psychiatry 2013; 73: 1180–1188.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME . Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995; 270: 1326–1331.
Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ . Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci 2000; 20: 7602–7613.
Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M . JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell 2003; 4: 521–533.
Björkblom B, Ostman N, Hongisto V, Komarovski V, Filén JJ, Nyman TA et al. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci 2005; 25: 6350–6361.
Oliva AA, Atkins CM, Copenagle L, Banker GA . Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci 2006; 26: 9462–9470.
Westerlund N, Zdrojewska J, Padzik A, Komulainen E, Björkblom B, Rannikko E et al. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat Neurosci 2011; 14: 305–313.
Komulainen E, Zdrojewska J, Freemantle E, Mohammad H, Marchisella F, Hollos P et al. JNK1 regulates dendrite architecture in the motor cortex and alters fine motor function. Front Cell Neurosci 2014; 8: 272.
Coffey ET . Nuclear and cytosolic JNK signalling in neurons. Nat Rev Neurosci 2014; 15: 285–299.
Baptista J, Mercer C, Prigmore E, Gribble SM, Carter NP, Maloney V et al. Breakpoint mapping and array CGH in translocations: comparison of a phenotypically normal and an abnormal cohort. Am J Hum Genet 2008; 82: 927–936.
McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 2009; 41: 1223–1227.
Pavlowsky A, Gianfelice A, Pallotto M, Zanchi A, Vara H, Khelfaoui M et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr Biol 2010; 20: 103–115.
de Anda FC, Rosario AL, Durak O, Tran T, Gräff J, Meletis K et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci 2012; 15: 1022–1031.
Winchester CL, Ohzeki H, Vouyiouklis DA, Thompson R, Penninger JM, Yamagami K et al. Converging evidence that sequence variations in the novel candidate gene MAP2K7 (MKK7) are functionally associated with schizophrenia. Hum Mol Genet 2012; 21: 4910–4921.
Marchisella F, Coffey ET, Hollos P . Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton (Hoboken) 2016; 73: 596–611.
van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH . Functional neurogenesis in the adult hippocampus. Nature 2002; 415: 1030–1034.
Smolinsky AM, Bergner CL, LaPorte JL, Kalueff AV Analysis of grooming behavior and its utility in studying animal stress, anxiety and depression. In: Gould TD (ed). Mood and Anxiety Related Phenotypes in Mice vol. 42. Humana Press: New York, NY, USA, 2009, pp 1–20.
Kuhn HG, Dickinson-Anson H, Gage FH . Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 1996; 16: 2027–2033.
Krishnan V, Berton O, Nestler E . The use of animal models in psychiatric research and treatment. Am J Psychiatry 2008; 165: 1109.
Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS . Hippocampal volume reduction in major depression. Am J Psychiatry 2000; 157: 115–118.
Frodl T, Carballedo A, Frey EM, O'Keane V, Skokauskas N, Morris D et al. Expression of glucocorticoid inducible genes is associated with reductions in cornu ammonis and dentate gyrus volumes in patients with major depressive disorder. Dev Psychopathol 2014; 26 (4 Pt 2): 1209–1217.
Wu MV, Hen R . Functional dissociation of adult-born neurons along the dorsoventral axis of the dentate gyrus. Hippocampus 2014; 24: 751–761.
Sun H, Kennedy PJ, Nestler EJ . Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 2013; 38: 124–137.
Myers AK, Meechan DW, Adney DR, Tucker ES . Cortical interneurons require Jnk1 to enter and navigate the developing cerebral cortex. J Neurosci 2014; 34: 7787–7801.
Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF . Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 2001; 50: 77–82.
Lee JK, Park J, Lee YD, Lee SH, Han PL . Distinct localization of SAPK isoforms in neurons of adult mouse brain implies multiple signaling modes of SAPK pathway. Brain Res Mol Brain Res 1999; 70: 116–124.
Doetsch F, García-Verdugo JM, Alvarez-Buylla A . Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci USA 1999; 96: 11619–11624.
Jan YN, Jan LY . Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci 2010; 11: 316–328.
Henze DA, Urban NN, Barrionuevo G . The multifarious hippocampal mossy fiber pathway: a review. Neuroscience 2000; 98: 407–427.
McAllister AK . Cellular and molecular mechanisms of dendrite growth. Cereb Cortex 2000; 10: 963–973.
Courtney MJ, Coffey ET . The mechanism of Ara-C-induced apoptosis of differentiating cerebellar granule neurons. Eur J Neurosci 1999; 11: 1073–1084.
Zhao C, Teng EM, Summers RG, Ming GL, Gage FH . Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci 2006; 26: 3–11.
Tararuk T, Ostman N, Li W, Björkblom B, Padzik A, Zdrojewska J et al. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol 2006; 173: 265–277.
Kempermann G . Seven principles in the regulation of adult neurogenesis. Eur J Neurosci 2011; 33: 1018–1024.
Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA . The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999; 22: 667–676.
Ben-Zvi A, Yagil Z, Hagalili Y, Klein H, Lerman O, Behar O . Semaphorin 3A and neurotrophins: a balance between apoptosis and survival signaling in embryonic DRG neurons. J Neurochem 2006; 96: 585–597.
Scharfman HE . The CA3 "backprojection" to the dentate gyrus. Prog Brain Res 2007; 163: 627–637.
Vivar C, Potter MC, Choi J, Lee JY, Stringer TP, Callaway EM et al. Monosynaptic inputs to new neurons in the dentate gyrus. Nat Commun 2012; 3: 1107.
Lacefield CO, Itskov V, Reardon T, Hen R, Gordon JA . Effects of adult-generated granule cells on coordinated network activity in the dentate gyrus. Hippocampus 2012; 22: 106–116.
Niwa H, Yamamura K, Miyazaki J . Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991; 108: 193–199.
Zhao C. Retrovirus-mediated cell labeling. In: Fred Gage GK, Song Hongjun (eds). Adult Neurogenesis. Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008, pp 101–117.
Yu DX, Di Giorgio FP, Yao J, Marchetto MC, Brennand K, Wright R et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep 2014; 2: 295–310.
Björkblom B, Vainio JC, Hongisto V, Herdegen T, Courtney MJ, Coffey ET . All JNKs can kill, but nuclear localization is critical for neuronal death. J Biol Chem 2008; 283: 19704–19713.
Charalampopoulos I, Vicario A, Pediaditakis I, Gravanis A, Simi A, Ibáñez CF . Genetic dissection of neurotrophin signaling through the p75 neurotrophin receptor. Cell Rep 2012; 2: 1563–1570.
Repici M, Centeno C, Tomasi S, Forloni G, Bonny C, Vercelli A et al. Time-course of c-Jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience 2007; 150: 40–49.
Yang SH, Sharrocks AD, Whitmarsh AJ . Transcriptional regulation by the MAP kinase signaling cascades. Gene 2003; 320: 3–21.
Jin J, Maren S . Prefrontal-hippocampal interactions in memory and emotion. Front Syst Neurosci 2015; 9: 170.
Tannenholz L, Jimenez JC, Kheirbek MA . Local and regional heterogeneity underlying hippocampal modulation of cognition and mood. Front Behav Neurosci 2014; 8: 147.
Maguire EA, Gadian DG, Johnsrude IS, Good CD, Ashburner J, Frackowiak RS et al. Navigation-related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci USA 2000; 97: 4398–4403v.
Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 2011; 472: 466–470.
Andreescu C, Mennin D, Tudorascu D, Sheu LK, Walker S, Banihashemi L et al. The many faces of anxiety-neurobiological correlates of anxiety phenotypes. Psychiatry Res 2015; 234: 96–105.
Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 2003; 9: 1180–1186.
Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G et al. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci 2005; 21: 363–377.
Sherrin T, Blank T, Hippel C, Rayner M, Davis RJ, Todorovic C . Hippocampal c-Jun-N-terminal kinases serve as negative regulators of associative learning. J Neurosci 2010; 30: 13348–13361.
Akers KG, Martinez-Canabal A, Restivo L, Yiu AP, De Cristofaro A, Hsiang HL et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 2014; 344: 598–602.
Thierry AM, Gioanni Y, Dégénétais E, Glowinski J . Hippocampo-prefrontal cortex pathway: anatomical and electrophysiological characteristics. Hippocampus 2000; 10: 411–419.
Satpute AB, Mumford JA, Naliboff BD, Poldrack RA . Human anterior and posterior hippocampus respond distinctly to state and trait anxiety. Emotion 2012; 12: 58–68.
Qi AQ, Qiu J, Xiao L, Chen YZ . Rapid activation of JNK and p38 by glucocorticoids in primary cultured hippocampal cells. J Neurosci Res 2005; 80: 510–517.
Mukherjee PK, DeCoster MA, Campbell FZ, Davis RJ, Bazan NG . Glutamate receptor signaling interplay modulates stress-sensitive mitogen-activated protein kinases and neuronal cell death. J Biol Chem 1999; 274: 6493–6498.
Thomas GM, Lin DT, Nuriya M, Huganir RL . Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. EMBO J 2008; 27: 361–372.
Egeland M, Zunszain PA, Pariante CM . Molecular mechanisms in the regulation of adult neurogenesis during stress. Nat Rev Neurosci 2015; 16: 189–200.
Rogatsky I, Logan SK, Garabedian MJ . Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA 1998; 95: 2050–2055.
Adzic M, Djordjevic J, Djordjevic A, Niciforovic A, Demonacos C, Radojcic M et al. Acute or chronic stress induce cell compartment-specific phosphorylation of glucocorticoid receptor and alter its transcriptional activity in Wistar rat brain. J Endocrinol 2009; 202: 87–97.
Acknowledgements
We are grateful to Petteri Piepponen, Jaakko Kopra and Juho-Matti Renko at the University of Helsinki for training HM to carry out cannulation and mini-pump implant surgeries. We thank the Cell Imaging Core at Turku Centre for Biotechnology for providing devices and technical support. This work was funded by EU FP7 ITN 608346 r’BIRTH and Academy of Finland Grants 135090 and 255537 to ETC that supported HM, FM, E F and SO-M, and the Sigrid Juselius Foundation Grant to ETC that funded PH. In addition, we thank the Doctoral Network in Molecular Biosciences at Åbo Akademi University for funding HM and PH and the Finnish Graduate School of Neuroscience for funding EK. This research was funded in part by the National Institute on Aging, Intramural Research Program (to BDP and HvP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Mohammad, H., Marchisella, F., Ortega-Martinez, S. et al. JNK1 controls adult hippocampal neurogenesis and imposes cell-autonomous control of anxiety behaviour from the neurogenic niche. Mol Psychiatry 23, 362–374 (2018). https://doi.org/10.1038/mp.2016.203
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.203
This article is cited by
-
HSPA12A controls cerebral lactate homeostasis to maintain hippocampal neurogenesis and mood stabilization
Translational Psychiatry (2023)
-
Stress-Activated Protein Kinase JNK Modulates Depression-like Behaviors in Mice
Molecular Neurobiology (2023)
-
JNK1 and JNK3: divergent functions in hippocampal metabolic-cognitive function
Molecular Medicine (2022)
-
A novel genomic region on chromosome 11 associated with fearfulness in dogs
Translational Psychiatry (2020)
-
Drug-responsive autism phenotypes in the 16p11.2 deletion mouse model: a central role for gene-environment interactions
Scientific Reports (2020)
