
Abstract
Stressful life events produce a state of vulnerability to depression in some individuals. The mechanisms that contribute to vulnerability to depression remain poorly understood. A rat model of intense stress (social defeat (SD), first hit) produced vulnerability to depression in 40% of animals. Only vulnerable animals developed a depression-like phenotype after a second stressful hit (chronic mild stress). We found that this vulnerability to depression resulted from a persistent state of oxidative stress, which was reversed by treatment with antioxidants. This persistent state of oxidative stress was due to low brain-derived neurotrophic factor (BDNF) levels, which characterized the vulnerable animals. We found that BDNF constitutively controlled the nuclear translocation of the master redox-sensitive transcription factor Nrf2, which activates antioxidant defenses. Low BDNF levels in vulnerable animals prevented Nrf2 translocation and consequently prevented the activation of detoxifying/antioxidant enzymes, ultimately resulting in the generation of sustained oxidative stress. Activating Nrf2 translocation restored redox homeostasis and reversed vulnerability to depression. This mechanism was confirmed in Nrf2-null mice. The mice displayed high levels of oxidative stress and were inherently vulnerable to depression, but this phenotype was reversed by treatment with antioxidants. Our data reveal a novel role for BDNF in controlling redox homeostasis and provide a mechanistic explanation for post-stress vulnerability to depression while suggesting ways to reverse it. Because numerous enzymatic reactions produce reactive oxygen species that must then be cleared, the finding that BDNF controls endogenous redox homeostasis opens new avenues for investigation.
Similar content being viewed by others


Introduction
Major stressful life events (for example, divorce, unemployment or the death of a relative) do not necessarily precipitate depression, but they can leave a trace, the allostatic load,1, 2 that creates a state of vulnerability to depression. Depending upon the level of vulnerability, subsequent stressful events may trigger depression.3 Treating vulnerable individuals during the prodromal phase could be highly beneficial and easier to do than after the onset of depression. Treating vulnerability to depression requires the knowledge of its underlying mechanisms, which may be different from those already identified for depression itself. To achieve this aim, it is necessary to have access to (i) two populations with different allostatic loads (but not displaying depression) and (ii) a biomarker to distinguish between the two populations. Experimental procedures have been designed to produce different allostatic loads in rodents. Social defeat (SD), which has been used as a major stressful event,4, 5, 6 can induce this situation.4, 5 SD, as a first hit, generates a state of vulnerability to depression in more than 40% of SD exposed Sprague-Dawley rats (vulnerable, V), while the rest of the population is identified as non-vulnerable (NV), since it is only after chronic mild stress (CMS—second hit), a month following SD, that V, but not NV, animals will display depression-like behavior.5 Hence, following a SD, V animals have a higher allostatic load than NV animals. Importantly, low serum brain-derived neurotrophic factor (BDNF) levels reliably identify V animals after the first hit5 as reported in human.7, 8, 9 This experimental protocol thus provides an ideal situation to study the mechanisms of vulnerability to depression since we have access to two populations with different allostatic loads and to a predictive biomarker at a stage when none of the animals express a depression-like phenotype (that is, after SD, but before CMS).
We hypothesized that oxidative stress would be a central mechanism to vulnerability to depression because hippocampal CA3 pyramidal cells display permanent dendritic retraction and spine loss in V animals,5 a typical signature of oxidative stress.10 In keeping with this hypothesis, stressful life events generate a high level of oxidative stress.11, 12, 13 Oxidative stress is the result of an imbalance between oxidant production and antioxidant mechanisms that leads to oxidative damage, including lipid peroxidation and DNA oxidation.14 A number of detoxifying (antioxidant) enzymes, including Cu-Zn superoxide dismutase (Cu-Zn SOD), peroxiredoxins (Prxs), catalase and glutathione peroxidases (GPxs), prevent such damage by limiting the accumulation of reactive oxygen species (ROS).15, 16 On the basis our hypothesis, we can make three predictions. First, one or several of these detoxifying/antioxidant enzymes are dysfunctional in V animals, thus generating oxidative stress. Second, the mechanisms regulating these enzymes are downregulated. Among these mechanisms, we focused on the Nrf2/Keap1 pathway, which is a master regulator of redox homeostasis.17, 18 Nrf2 is usually retained in the cytosol, where it is tethered to its cytosolic repressor, Kelch-like ECH associated protein-1 (Keap1).17, 18 When Nrf2 dissociates from Keap1, it translocates to the nucleus and binds to antioxidant response elements on target genes to activate antioxidant defenses.18, 19 This redox-sensitive transcription factor controls the expression of sulfiredoxin,20 an important regulator of peroxiredoxin activity,21 which catalyzes the Prx-2-SO2H reduction, thus regenerating the active enzyme, Prx-2.18, 22 Our choice of Nrf2 is justified because its dysfunction produces a redox imbalance in neurodegenerative, cardiovascular, pulmonary and chronic inflammation,19, 23 which can lead to a depression-like phenotype.24, 25 The third prediction is that BDNF, which levels remain low in V animals, is causally related to oxidative stress.
Here, using a longitudinal study, we demonstrate that low BDNF levels prevent the activation of Nrf2-dependent endogenous antioxidant peroxiredoxin/sulfiredoxin defense mechanisms, leading to sustained oxidative stress in vulnerable animals. Reactivating these defense mechanisms restores redox homeostasis and abolishes vulnerability to depression.
Materials and methods
Animals
Male Sprague-Dawley rats weighing 290–310 g (9 weeks old at the beginning of the experiments) were obtained from the Centre d'Elevage R. Janvier (53940 Le Genest-St-Isle, France) and used as intruder rats. We used male Sprague-Dawley rats from the same litter. From each litter, some rats were selected randomly as controls, and the others were subjected to repeated SD (see below).
Male wild-type Groningen (WTG) rats were used as resident rats.26
Wild-type (Nrf2+/+) and Nrf2-null (Nrf2−/−) mice were used to confirm the role of Nrf2. Nrf2-null mice were backcrossed onto the C57BL/6J background for seven generations using alternating male and female stock mice from the CNRS TAAM UPS44 (Orléans). The resulting wild-type (Nrf2+/+) and Nrf2-null (Nrf2−/−) mice were genotyped and only male mice at 6 weeks old at the beginning of the experiments were used. They were exposed to CMS only.
All animals (rats or mice) were housed in chronobiological animal facilities equipped with regularly spaced, soundproof, temperature-controlled compartments and supplied with filtered air. Procedures involving animals and animal care protocols were performed in accordance with institutional guidelines that conformed to national and international laws and policies. For more details, please see the information in the Supplementary Materials and Methods.
Social defeat procedure in rats
The social defeat procedure (SD) (D-3 to D0) was performed on rats as previously described.5, 27, 28 This procedure involved subjecting the same pairs of residents and intruders to four daily sessions (Figure 1a). The 45 min sessions were divided into two consecutive periods. During period I (30 min), intruders were placed individually in a protective cage within the resident animal’s home cage. The protective cage allowed unrestricted visual, auditory and olfactory contact with the resident but prevented close physical contact. During period II (15 min), the protective cage was removed, either with the resident remaining present, which allowed for physical confrontation with the intruder (3–4 confrontations of ~10 s, during each of which the intruding (defeated) animal was always dominated by the resident rat) or with the resident removed, which gave the intruder access to the entire resident home cage (control intruders). The control intruders were therefore never physically attacked and defeated by the resident and did show any sign of stress and/or anxiety.27, 28 Animals that were wounded as a result of the SD procedure were excluded from the experiment (2% of stressed animals).

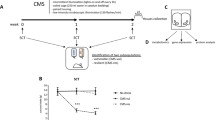
Oxidative stress in vulnerability to depression. Restoration of redox homeostasis and reversion of the phenotype of vulnerability with antioxidant treatment. (a) The experimental 'double-hit' design involved subjecting rats to SD (D-3-D0) followed, 4 weeks later, by CMS. Evaluation of redox parameters was performed 5 days (D5), 11 days (D11) and 4 weeks (D31) after the end of the social defeat procedure. Neuroanatomical parameters were measured at D11 and D31. Depression-like phenotype (helplessness behavior, decrease in sweet water consumption, HPA, body weight) was evaluated at D31 and at the end of the double-hit procedure, at D53. Animals were treated with 7,8-DHF or with Tempol: 1- for 7,8-DHF treatment, administrations were performed at D5, D7 and D9; 2- a 15-day-long treatment with Tempol started 11 days after SD (D11). (b) Oxidative stress results in lipid oxidative damage (lipid peroxidation, MDA) and in decreased activity of the sensitive redox aconitase in vulnerable (V, n=6–8) rats, but not in NV (n=7–8) animals, 31 days after the end of the SD procedure (D31). (c) The quantity and activity of SOD, which catalyzes the ROS hydrogen peroxide (H2O2) production and of the antioxidant systems involved in the reduction of ROS H2O2 to water, peroxiredoxin-2 (Prx-2) and its inactive form (Prx-sulfinic [SO2]/sulfonic [SO3]), glutathione peroxidase (GPx) and catalase activities, were evaluated in V animals and NV animals. The peroxiredoxin-2 antibody targets both the active and inactive forms of the enzyme (total). The Prx-SO2/SO3 antibody targets the inactive form of enzyme. SOD activity and the inactive form of Prx-2 were increased in V animals indicating oxidative stress. (d) Effects of a 15-day-long treatment with Tempol starting 11 days after SD (D11), when vulnerable (V) could be identified based on serum BDNF levels. Tempol normalized all signs of oxidative stress in V animals to control and NV levels. Tempol prevented lipid oxidative damage, decrease in aconitase, increase in SOD, and restored the active form of peroxiredoxin-2 (Prx-2). Glutathione peroxidase (GPx) and catalase activities were not affected (C: n=5–6; NV: n=6–8; V: n=5–6). (e) Tempol restored anatomical integrity (total apical dendrite length and spine density) of CA3 hippocampal neurons in V animals (n=4–5). (f) Tempol prevented the appearance of a depression-like phenotype of V animals after CMS as assessed by immobility time in the forced swim test, sweet water consumption and activity of HPA axis (C: n=6–8; NV: n=10; V: n=7–9). See also Supplementary Figure S1d for proteomic analysis 31 days after SD. The results of statistical analyses are presented in Supplementary Table S1A1-2. *P<0.05 vs C animals; aP<0.05 vs NV group; †P<0.05 vs the corresponding Tempol-treated group. BDNF, brain-derived neurotrophic factor; CMS, chronic mild stress; DHF, dihydroxyflavone; HPA, hypothalamic–pituitary–adrenal hyperactivity; NV, non-vulnerable; ROS, reactive oxygen species; SD, social defeat; SOD, superoxide dismutase.
Serum BDNF assay
Blood samples (200 μl) were collected from conscious rats between 1200 and 1230 hours at key times (D-4, D5, D11 and D31). These samples were taken from the tail vein and were collected in Eppendorf tubes.5 The samples were centrifuged to separate the serum, which was stored at −20 °C until BDNF analysis. BDNF concentrations were determined at a dilution of 1:25 with a commercial BDNF assay (Promega, Charbonnières-les-Bains, France). The measurement of serum BDNF levels was performed after all other experiments and analyses were performed. Thus, the investigators were blind to the classification of animals based on BDNF level, which was the last parameter to be measured during the experimental procedure and analyses.
Proteomic approach
To investigate proteins related to redox homeostasis, we analyzed hippocampal extracts in a urea-based solubilization buffer with comparative two-dimensional (2D) gel analysis. Spots displaying differential expression were identified using MALDI-TOF MS. Detailed descriptions of our 2D gel electrophoresis and identification protocols are provided in the Supplementary Materials and Methods.
Redox parameters
Catalase, SOD, glutathione peroxidase, aconitase and TBARS levels were immediately analyzed in fresh hippocampal extracts. Detailed descriptions of extraction and assay protocols are provided in the Supplementary Materials and Methods.
Immunofluorescence staining, confocal microscopy and image analysis
Oxidative damages were visualized in all subfields of the dorsal and ventral hippocampus using a DNA adduct formed by the reaction of OH radicals with the DNA base guanine.29 Detailed descriptions of immunofluorescence staining protocols, confocal microscopy and image analysis are provided in the Supplementary Materials and Methods.
Golgi staining
Neuroanatomical alterations were analyzed in unperfused brains using the Golgi staining method following the manufacturer’s instructions. The total length of the apical dendrites and the density of apical dendritic spines of selected neurons in CA3 were determined with AxioVision 4.7 software (Marly Le Roi, France) and a Zeiss Imager M1 microscope (Marly Le Roi, France). Detailed descriptions of the dendrite measurement protocols, Sholl analysis and spine density assessments are provided in the Supplementary Materials and Methods.
Western blot analysis
After nuclear and cytosolic fractionation of hippocampal tissues (extensive details are included in the Supplementary Materials and Methods), proteins were resolved by SDS–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes and incubated with antibodies specific to SOD1, Prx-2, Prx-SO2/SO3, Nrf2, Keap1, aconitase, GAPDH or β-actin. The secondary antibodies used in these assays were either IRDye- or horseradish peroxidase-conjugated (for further information, please see the Supplementary Materials and Methods).
RT–PCR
After RNA extraction, real-time PCR amplification was performed to analyze the following target genes: sulfiredoxin (Rn01536084-g1*) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Rn99999916-S1). Descriptions of RNA extraction protocols and real-time PCR amplification conditions are provided in the Supplementary Materials and Methods.
In vivo siRNA knockdown, microinfusion and ICV administration
BDNF knockdown was performed using Silencer Select Ambion in vivo siRNA targeting BDNF (with additional chemical modifications for superior stability, Invitrogen, Courtaboeuf, France) or a non-targeting Silencer Select Ambion in vivo siRNA as a control (Invitrogen). A week before siRNA transfections, rats were anesthetized with ketamine (33 mg kg−1) and xylazine (6 mg kg−1) and were placed in a stereotaxic apparatus (Kopf Instruments, Phymep, Paris, France). Two stainless steel cannulas (Plastic One, Phymep, Paris, France) were implanted bilaterally in the dorsal hippocampus at the following stereotaxic coordinates with respect to bregma: −4.8 mm anteroposterior, ±3.2 mm mediolateral and −2.2 mm dorsoventral (from dura mater, histological location of canula was verified in two sham animals, see Supplementary Figure S1a). The cannulas were anchored to the skull using acrylic dental cement. Control siRNA and BDNF siRNA (final concentration of 50 μm, Ambion—48 h stability—, InVitrogen) were prepared using Invivofectamine 2.0 according to the manufacturer’s forward protocol (Invitrogen). Animals received two injections of control or BDNF siRNA 2 days apart and were killed 48 h later. Each hippocampus received 1 μl of solution at a flow rate of 0.25 μl min−1. The injection needle was left in situ for 5 min post injection and then slowly removed. The brain of the animals was removed and the hippocampi were dissected to obtain nuclear and cytosolic extracts after subcellular fractionation. BDNF siRNA efficiency was 62%: BDNF levels were 5.73±0.17 pg mg−1 with control siRNA and 2.17±0.06 pg mg−1 with BDNF siRNA.
For microinjection of BDNF, rats were anesthetized with ketamine and xylazine. Two stainless steel cannulas (Plastic One) were implanted in each dorsal hippocampus (as above). Infusions of 1 μg BDNF, or of vehicle, were performed during 1 h. Subcellular fractionation was performed as above.
For intracerebroventricular delivery with osmotic minipumps, we prepared tert-Butylhydroquinone (t-BHQ, Sigma) at a final concentration of 1 mm in vehicle (1% EtOH in H2O).30 Alzet osmotic minipumps (Charles River Laboratories, L’Arbresle, France) were filled with a fresh solution of t-BHQ and were connected to a 26-gauge stainless steel cannula implanted in the right lateral ventricle (stereotaxic coordinates with respect to bregma: 1 mm caudal; −1.5 mm lateral; −3.4 mm below the surface) for continuous infusion during 6 (t-BHQ) or 7 (vehicle) days. Administration of t-BHQ started 6 days before the onset of the CMS protocol.
Superfusion of the hippocampus
Hippocampi were cut into eight pieces, placed in the tissue chamber for perfusion, and suspended in artificial cerebrospinal fluid (pH 7.3). After 1 h of superfusion,31 the tissue was collected and subjected to subcellular fractionation. The details of the superfusion procedure are provided in the Supplementary Materials and Methods.
Two-hit procedure
Sprague-Dawley rats were subjected to SD (first hit), and then, 4 weeks later, to 3 weeks of CMS (second hit) (Figure 1a). The CMS protocol was performed as previously described.5 The control rats (C rats) were control intruders (not subjected to SD) that were not exposed to CMS.
Chronic mild stress protocol
The CMS protocol is described in the Supplementary Materials and Methods.
Phenotypic analyses
The depressive phenotype of the animals was determined by analyzing immobility time in the forced swimming test, the amount of liquid consumed in the sweet water consumption test, the activity of the HPA axis (serum corticosterone levels and adrenal gland weight) and body weight. Detailed protocols used in the forced swimming and sweet water consumption tests and to acquire measurements of HPA axis activity are extensively described in the Supplementary Materials and Methods.
Drugs and treatments
The antioxidant compound, Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl; 288 μmol kg−1 day−1),32, 33 was dissolved in distilled water and delivered via ALZET osmotic minipumps. 7-8-dihydroxyflavone (7,8-DHF; 5 mg kg−1)34 was dissolved in sterile 0.9% NaCl solution adjusted to pH 7 with 0.1 m NaOH and administered (i.p.) on days 5, 7 and 9 after the end of the SD protocol (D5, D7 and D9). For ICV administration, a prototypical compound that induces the nuclear translocation of Nrf2, t-BHQ (1 mm),18, 30 was dissolved in 1% EtOH/distilled water and delivered via ALZET osmotic minipumps. For superfusion experiments, 7,8-DHF (10−5 m) was diluted in artificial cerebrospinal fluid. For more details regarding the drugs and treatments used, please see the Supplementary Materials and Methods.
Statistical analyses
Classification of animals into NV or V was performed using the k-means clustering method with Statistica.35 Differences in SOD activity, TBARS levels, aconitase activity or quantity, glutathione peroxidase activity, catalase activity, peroxiredoxin-2, peroxiredoxin SO2/SO3, the marker 8-oxoDG, the gene expression of nuclear Nrf2, cytosolic Nrf2, cytosolic Keap1, and sulfiredoxin, immobility time, anhedonia, HPA axis activity, hippocampal BDNF levels, and the apical dendrite length of CA3 neurons and hippocampal CA3 dendritic spines were evaluated by one-way analysis of variance (ANOVA). Serum BDNF concentrations or body weight were analyzed by one-way ANOVA for repeated measures. Stress effects and treatment effects were evaluated by two-way ANOVA. All results from ANOVA analyses (F-values and P-values) are provided in Supplementary Tables 1 and 2. When an ANOVA revealed a significant effect, a pair-wise post hoc Bonferroni test was performed. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally employed in comparable studies. All data are presented as the mean±s.e.m.
Results
Oxidative stress in vulnerable animals
If oxidative stress contributes to vulnerability to depression, markers of oxidative stress should be present in V but not in NV rats. Hippocampal tissues from both groups were analyzed thirty-one days after SD (D31), when V and NV animals can be distinguished based on their serum BDNF levels5 and do not present anymore an anxious or a depression-like phenotype (Supplementary Figure S1b,c).5 We found lipid oxidative damage (indicated by increased lipid peroxidation) and a decrease in the activity of aconitase, an intracellular enzyme highly sensitive to pro-oxidant conditions,36 specifically in V animals (Figure 1b). Two-dimensional gel profiling revealed differences in the expression of eleven abundant proteins that were clustered into four main functional categories according to in silico analysis with GO Term Finder37 (Supplementary Figure S1d). One category included SOD and Prx-2 and Prx-6, which are enzymes involved in the regulation of ROS levels. Consistently with this proteomic approach, subsequent biochemical analyses showed that the quantity and activity of SOD, an inducible enzyme that catalyzes the reduction of the ROS superoxide radical (O2•-) to the ROS hydrogen peroxide (H2O2),38 were higher in V than in NV animals (Figure 1c). Prx-2, which is part of a major enzymatic system that is involved in the reduction of the ROS H2O2 to water, was present primarily in its inactive form, Prx-sulfinic/sulfonic (SO2/SO3), in the hippocampi of V animals. However, there was no significant change in its overall level of expression (Figure 1c). In contrast, the activity of both GPx and catalase remained unchanged in V and NV animals (Figure 1c). These results suggest that an increase in the inactive form of Prx-2 leads to defective H2O2 clearance in the hippocampi of V animals (thereby inducing oxidative stress). The levels of H2O2 could not be measured directly, because H2O2 cannot be accurately quantified in ex vivo brain material.39 If oxidative stress is causally related to vulnerability to depression, antioxidant treatment should reverse this vulnerability phenotype.
Vulnerability to depression after SD is due to oxidative stress
We used Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidinoxyl), an antioxidant compound that promotes the metabolism of many ROS33 to treat V and NV rats. As expected, Tempol reduced GPx levels33 and fully reestablished redox homeostasis in V animals (Figure 1d). We have previously shown that both the length of apical, but not basal, CA3 pyramidal cell dendrites and the number of spines are consistently decreased in V animals.5 Since such alterations can sign cellular oxidative stress,40 we used them as anatomical markers. Tempol treatment fully normalized the lenght of apical dendrites and the number of spines to control levels (Figure 1e and Supplementary Figure S1e). Finally, Tempol abolished vulnerability to depression. Increased immobility times in the forced swimming test, decreased sweet water consumption and HPA axis hyperactivity (corticosterone levels, adrenal gland weight) observed in vehicle-treated V animals were prevented by Tempol treatment in V animals, following the second hit (Figure 1f). In NV animals, CMS protocol induced a small increase of corticosterone levels5 that was not prevented by Tempol treatment. Together, these results demonstrate that, in this double-hit experimental model, oxidative stress is responsible for the vulnerability to depression, and that such vulnerability can be reversed with an antioxidant treatment.
Longitudinal study of oxidative stress status
Oxidative stress was identified at 31 days after SD in V animals, which suggests two possible scenarios: (i) a gradual buildup of oxidative stress or (ii) a long-lasting state triggered by the first hit. We thus performed a longitudinal study to distinguish between these two possibilities. A state of oxidative stress was present in all animals at D5 (Supplementary Figure S2a), which is a classical characterization of an acute response to an intense stressor.12, 13 We then assessed oxidative stress at D11, when V and NV animals can first be distinguished based on serum BDNF levels (Figure 2a). Animals identified as V at D11 remained V at D31 (Figure 2a), whereas control (C) animals maintained constant levels of BDNF throughout. On D11, V animals displayed increased SOD activity, enhanced levels of the inactive form of Prx-2 (Figure 2b) and decreased aconitase activity (Supplementary Figure S2b), which should result in H2O2 accumulation and oxidative stress. A state of oxidative stress was directly confirmed by the presence of high levels of lipid peroxidation (MDA) and DNA oxidation (using 8-oxo-dG as a marker) in the hippocampus of vulnerable animals (Figures 2c and d). Oxidative stress in vulnerable animals was mostly found in neurons (Figure 2e). In contrast, SOD activity returned to normal, levels of the inactive form of Prx-2 decreased and oxidative damage disappeared by D11 in NV animals (Figures 2b–d and Supplementary Figure S2b). From these results, we conclude that the acute oxidative stress response triggered by the first hit was maintained in V animals but disappeared in NV animals. Because Prx-2 activity has a key role in maintaining redox homeostasis,41 we next explored the mechanisms underlying its long-term inactivation.

Dysfunction of the redox-sensitive transcription factor Nrf2 in vulnerable animals. (a) Longitudinal follow-up of serum BDNF levels leading to the identification of NV and V rats already 11 days after SD (D11). (b) At D11, only V animals showed increased SOD activity and increase in the inactive form of Prx-2 (C: n=5–6; NV: n=6–8; V: n=5–6), as at D31. (c and d) Oxidative damages estimated with lipid peroxidation [MDA, c] and DNA oxidation [8-oxo-dG marker, d] in ventral and dorsal hippocampi of NV (n=5–8), V (n=4-6) and control (n=5–6) animals at D11. MDA analysis also revealed increased levels of lipid peroxidation in V animals. Upper panel: Immunofluorescence micrographs illustrating specific DNA oxidation in all subfields of the ventral hippocampus of V animals as compared with NV and controls (Dentate Gyrus: DG, CA3, CA1). Scale bars, 100 μm. (lower) Quantitative analysis showing DNA damage in ventral and dorsal hippocampi of V animals. DNA damage was specifically observed in V animals. (e) 1- Pictomicrograph of co-immunolabeling for NeuN (blue), GFAP (red) and 8-oxo-dG (green) in V animals. 2- Pictomicrograph with increased magnification. V rats showed 8-oxo-dG (green) immunolabeling in neurons (NeuN, blue) and some glial (GFAP, red) cells. Scale bar: 30 μm. (f) The levels of sulfiredoxin mRNA—an adjunct of peroxiredoxin system regenerating the active enzyme—were increased in NV but decreased in V animals as compared with controls. Nuclear levels of Nrf2 were increased in NV but decreased in V animals as compared with controls (C: n=7, NV: n=7; V: n=6). These results suggest activation of antioxidant defense mechanisms in NV and their failure to activate in V animals. (g) The levels of sulfiredoxin mRNA and nuclear Nrf2 in hippocampi were decreased in animals at 5 days after SD. (SD, n=6) and control animals (C, n=6). The results of statistical analyses are presented in Supplementary Table S1 B1-2. *P<0.05 vs C animals; aP<0.05 vs NV group. See also Supplementary Figure S2 for oxidative stress; cytosolic Nrf2 and Keap1 in animals 5 and 11 days after SD; microglia and phagocytic microglial immunolabeling; repeated social stress in mice; t-BHQ effect; hippocampal BDNF levels 5 and 31 days after SD and Supplementary Table S2 B1-5 for statistical analysis. BDNF, brain-derived neurotrophic factor; NV, non-vulnerable; SD, social defeat; SOD, superoxide dismutase.
Dysfunction of the Nrf2 pathway causes vulnerability to depression
The active form of Prx-2 is regenerated by sulfiredoxin.21 Sulfiredoxin mRNA levels were decreased in V animals and increased in NV animals compared with controls on D11 (Figure 2f), suggesting that transcriptional downregulation of sulfiredoxin contributed to the accumulation of the inactive form of Prx-2 in V animals, whereas its upregulation in NV animals contributed to detoxification. The transcription of sulfiredoxin is controlled by the redox-sensitive transcriptional factor Nrf2,20, 22 a master regulator of redox homeostasis.17, 18 At D5, all experimental animals had lower nuclear levels of Nrf2 than controls (Figure 2g), in keeping with decreased levels of sulfiredoxin (Figure 2g), increased levels of the inactive form of Prx-2 (Supplementary Figure S2a), and a state of oxidative stress (Supplementary Figure S2a). At D11, nuclear Nrf2 levels were significantly increased in NV animals as compared with controls (Figure 2f), indicating the activation of antioxidant defenses. In contrast, Nrf2 nuclear levels remained decreased in V animals, which may have induced a permanent state of oxidative stress and vulnerability to depression in these animals. If the Nrf2 pathway is causally linked to vulnerability to depression, impairment of Nrf2 would produce vulnerability to depression. To test this hypothesis, we first evaluated the behavior of Nrf2+/+ mice in avoidance test after 10 days social stress predicting that Nrf2+/+ mice can also be split into two populations4 (see Supplementary Figure S2f). Secondly, we used Nrf2-null mice predicting that they would be endogenously vulnerable, that is, a minor stressor (CMS) should be sufficient to trigger a depression-like phenotype.
Nrf2 downregulation is sufficient to cause vulnerability to depression
Nrf2-null mice were constitutively characterized by oxidative stress (Figure 3a) and by anatomical alterations in CA3 pyramidal cells (Figure 3b). These were abolished by Tempol treatment (Figure 3b). Hippocampal BDNF levels were not significantly different in Nrf2-null mice compared with Nrf2+/+ mice (1.71±0.06 pg mg−1 tissue (n=6) compared with 2.12±0.26 pg mg−1 tissue (n=5), respectively). Nrf2-null mice did not display a depression-like phenotype. Their immobility times in the forced swim test and the activity of their HPA axes were similar to those observed in Nrf2+/+ mice (Figure 3c). Sweet water consumption was also similar between Nrf2+/+ and Nrf2-null mice (6.6 and 7.4 ml per day, respectively). However, when exposed to three weeks of CMS, Nrf2-null mice developed a depression-like phenotype that included helplessness behavior, anhedonia and HPA hyperactivity, all of which were prevented by pretreatment with the antioxidant Tempol (Figure 3c). Nrf2-null mice are thus endogenously vulnerable to depression, sharing the phenotypic traits of V rats (including an endogenous state of oxidative stress). The depressive phenotype of Nrf2-null mice can be revealed by exposure to a minor stressor (CMS). We conclude that the downregulation of Nrf2 is sufficient to cause vulnerability to depression.

Nrf2 alteration is necessary and sufficient to induce vulnerability to depression. (a) Lipid peroxidation (MDA) was increased in Nrf2-null mice (Nrf2−/−) as compared with Nrf2+/+ (n=6, for each). (b) Apical CA3 dendritic architecture was altered in Nrf2−/− mice. Alterations were reversed following a 4-week Tempol (Tp) treatment (n=6–7 mice). (c) Evaluation of depression-like phenotype as assessed with immobility time in the forced swim test, sweet water consumption and HPA axis activity of Nrf2−/− and Nrf2+/+ mice, treated or not with Tempol (Tp), submitted or not to 3 weeks of CMS (n=6-9 mice). Note that Nrf2−/− mice were endogeneously in a vulnerable state, as CMS was sufficient to induce a depression-like phenotype. In contrast, CMS did not trigger a depression-like phenotype in Nrf2+/+ mice. Tempol treatment fully reversed the vulnerability state of Nrf2−/− mice. (d) Treatment with the Nrf2 activator t-BHQ abolished the depression-like phenotype in V rats (n=6), as compared with vehicle-treated V rats (n=8). (e) In vivo injection of BDNF siRNA in rat hippocampus decreased Nrf2 nuclear levels as compared with control siRNA or saline (Sal) injections (n=5–6). Microinjection of siRNA BDNF triggered an oxidative stress response as assessed by lipid peroxidation. (f) In vivo microinjection of BDNF increased the translocation of Nrf2 to the nucleus compartment (n=4). (g) SD in rats decreased Nrf2 translocation to the compartment of nucleus. Hippocampal perfusion with 7,8-DHF (10 μm) increased Nrf2 translocation in control and SD rats (C: n=6–7; SD: n=6–7). The results of statistical analyses are presented in Supplementary Table S1 C1-5. *P<0.05 vs Nrf2+/+ mice, vs vehicle (Veh), vs siRNA control or vs C animals; aP<0.05 vs NV rats or vs saline (Sal) group; †P<0.05 vs the corresponding treated group. See also Supplementary Figure S3a,b for cytosolic Nrf2 in in vivo siRNA condition; cytosolic Nrf2 in ex vivo condition and Supplementary Table S2 C1-2 for statistical analysis. BDNF, brain-derived neurotrophic factor; CMS, chronic mild stress; DHF, dihydroxyflavone; HPA, hypothalamic–pituitary–adrenal hyperactivity; NV, non-vulnerable; ROS, reactive oxygen species; SD, social defeat.
Nrf2 downregulation is necessary to cause vulnerability to depression
If the downregulation of Nrf2 constitutes a necessary condition for vulnerability to depression, inducing its translocation should abolish vulnerability to depression. We thus used V and NV rats and administered t-BHQ, a prototypical compound that induces the nuclear translocation of Nrf218 (Supplementary Figure S2g). Treatment with t-BHQ abolished vulnerability to depression in V rats (Figure 3d). We conclude that altered Nrf2 function is necessary and sufficient for maintaining a long-lasting state of oxidative stress and vulnerability to depression.
Upstream regulation of Nrf2
In the double-hit scenario tested here, we demonstrated (i) that a sustained state of oxidative stress caused vulnerability to depression (since the latter could be reversed by the administration of detoxifying agents), and (ii) that Nrf2 dysfunction was responsible for the sustained state of oxidative stress (since inducing Nrf2 translocation reversed oxidative stress and the vulnerability state). We therefore investigated which upstream mechanisms may be responsible for the long-term downregulation of Nrf2. The candidate mechanism should follow the time course of Nrf2/oxidative stress alterations, that is, it should be altered during the acute phase after SD, it should persist in V animals, and it should be resolved in NV animals. In addition, detoxification (as a downstream effector), although restoring a control behavioral phenotype, may not necessarily affect this upstream mechanism. BDNF fulfills these requirements. Its serum levels, which reflect hippocampal BDNF concentrations,5, 42 showed the same time course as Nrf2/oxidative stress alterations (Figure 2a). In addition, BDNF levels were not modified following the restoration of redox homeostasis suggesting that oxidative stress does not affect BDNF levels (Supplementary Figure S2h). We thus explored the possibility that BDNF may directly regulate Nrf2.
BDNF controls the stability of the redox-sensitive Nrf2 pathway
If the hypothesis that BDNF directly regulates Nrf2 is correct, altering BDNF levels should modify the amount of Nrf2 translocation. This hypothesis was tested in vivo. Microinjections of BDNF siRNA in rat hippocampi decreased Nrf2 translocation to the nucleus and caused oxidative stress (Figure 3e). In contrast, no such modifications were induced by microinjections of control siRNA or saline (Figure 3e). Conversely, in vivo microinjection of BDNF into the rat hippocampi increased Nrf2 translocation to the nuclear compartment (Figure 3f). Since direct in vivo down- and upregulation of BDNF decreases and increases Nrf2 translocation, respectively, we propose the following putative molecular cascade to explain vulnerability to depression in the double-hit scenario studied here. Low BDNF levels prevent sufficient translocation of Nrf2 to the nucleus, resulting in decreased Prx-2/Sulfiredoxin-dependent detoxifying activity. This produces the oxidative stress state that is responsible for vulnerability to depression. If this scheme is correct, upregulating BDNF should abolish the vulnerability state of V animals.
This hypothesis could not be directly tested because of the unsuitable pharmacokinetic profile and adverse long-term side effects triggered by direct injections of BDNF.43 We thus tested 7,8-DHF, a flavone, which has been identified as a selective TrkB agonist, but which can act independtly as a potent antioxidant to protect cells against glutamate or H2O2-induced oxidative injury.44, 45, 46 Perfusion of hippocampal tissues extracted from control and SD rats with 7,8-DHF (10 μm) induced a marked increase in the translocation of Nrf2 to the nuclear compartment (Figure 3g). Since administration of 7,8-DHF can restore Nrf2 translocation, we predicted that treating V animals with 7,8-DHF would abolish their state of vulnerability.
Reversal of vulnerability to depression
Systemic injections of 7,8-DHF normalized Nrf2 translocation to the nuclear compartment in V animals to NV and control levels, restored normal levels of (i) sulfiredoxin gene expression (Figure 4a), (ii) the active form of Prx-2 (Figure 4b), (iii) DNA oxidation (Figure 4c) and (iv) lipid peroxidation (Supplementary Figure S3d). Treatment with 7,8-DHF also normalized anatomical alterations of CA3 pyramidal cells (Figure 4d). Finally, 7,8-DHF treatment in V animals abolished their vulnerability to depression since they did not display phenotypic traits of depression after CSD (Figure 4e). Thus, treatment with 7,8-DHF restored Nrf2 translocation, reinstated redox homeostasis and abolished vulnerability to depression.

Restoring BDNF-dependent Nrf2 nuclear translocation re-establishes redox homeostasis, repairs neuronal damage and abolishes vulnerability to depression. (a) Systemic injections of 7,8-DHF restored Nrf2 translocation to the nucleus and expression of sulfiredoxin in V rats (n=7) at D11, as compared with vehicle-treated V animals (n=6). (b) 7,8-DHF treatment restored the expression of the active form of Prx-2 in V animals as compared with vehicle-treated animals (C: n=5; NV: n=6–7; V: n=5–6). (c) Reversion of DNA oxidation [8-oxo-dG marker] by 7,8-DHF in the ventral and dorsal hippocampus of vulnerable (V, n=5) as compared with NV (n=5), and control (C, n=4) animals at D11. Immunofluorescence micrographs illustrating DNA oxidation in all subfields of the ventral hippocampus of treated, or not, control (C), NV and V animals (Dentate Gyrus: DG, CA3, CA1). Scale bars, 100 μm. (d) The total apical dendritic length of CA3 neurons and spine density was restored in 7,8-DHF-treated V and NV animals, as compared with vehicle-treated animals. (e) Treatment with 7,8-DHF in animals identified as V at D11 (n=6–7) completely prevented the occurrence of a depression-like phenotype following CMS (n=8). The results of statistical analyses are presented in Supplementary Table S1 D1-3. *P<0.05 vs C animals; aP<0.05 vs NV animals; †P<0.05 vs the corresponding 7,8-DHF-treated group. See also Supplementary Figure S3c-e for cytosolic Nrf2 or Keap1, neuroanatomical alterations and oxidative stress in non-vulnerable, vulnerable or control animals treated, or not, with 7,8-DHF, 11 days after the SD and Supplementary Table S2 C3-4 for statistical analysis. BDNF, brain-derived neurotrophic factor; CMS, chronic mild stress; DHF, dihydroxyflavone; NV, non-vulnerable; SD, social defeat.
Discussion
SD during the acute phase (5 days after the end of SD, Figure 5) produces an oxidative stress response that manifests as SOD hyperactivity and an increase in the inactive form of Prx-2, resulting in lipid damage. Prx-2 is an antioxidant enzyme involved in H2O2 reduction. Catalytic H2O2 reduction by this typical 2-Cys Prx involves the oxidation of its redox-active peroxidatic cysteine into a sulfenic acid (Cys-SOH), which can be recycled via the formation and reduction of an intermediate disulfide bond.41 Prx-2 is itself sensitive to inactivation by high levels of H2O2 through the oxidation of its peroxidatic cysteine to a sulfinic (–SO2H) or sulfonic (–SO3H) acid form.47 Thus, overoxidation of Prx-2 favors H2O2 accumulation and redox imbalance.48 Prx-2 is strongly expressed in neurons, especially in the hippocampus.41 Dysfunction of Prx-2 leads to oxidative stress and cell damage in neurodegenerative diseases.41, 49 The increase in the Prx-2 inactive form observed just after SD is consistent with the depleted antioxidant levels and altered antioxidant enzyme activities that have been measured in various acute or chronic models of stress, including models of immobilization stress, swim stress, CMS and SD.12, 13 This downregulation could be a compensatory response or an essential phase that protects against deleterious chronic stress-related events rather than a maladaptive response.50, 51 Following the initial acute phase of redox imbalance, oxidative stress disappeared in NV animals by D11 but persisted in V animals (Figure 5). This finding and the observation that the vulnerable state can be suppressed by treatment with antioxidants leads us to propose that an unresolved state of oxidative stress leads to vulnerability to depression in the double-hit scenario tested here. This scheme is similar to that proposed for inflammatory diseases, in which acute inflammatory processes need to be cleared to avoid a chronic state.52 Our results are consistent with the channel oxidation hypothesis of depression-like behaviors, which can be reversed by the administration of Tempol.53

Summary scheme for vulnerability to depression after SD. A major stressful event leads to an early oxidative stress response, which needs to be cleared by endogenous antioxidant processes. If oxidative stress persists, a vulnerability state to depression is produced. Low BDNF levels in SD animals, during the acute phase, impair appropriate Nrf2 translocation, and the transcription of the downstream genes, particularly sulfiredoxin involved in the peroxiredoxin-2 regeneration leading to oxidative stress. In NV animals, the return to baseline BDNF after SD enables normal Nrf2 function, with the activation of defense mechanisms, in particular, sulfiredoxin to restore Prx-2 activity and H2O2 clearance and a return to normal redox state. Restoration of redox homeostasis prevents the development of a depression-like phenotype after CMS. In vulnerable animals, persistent low BDNF levels impair appropriate Nrf2 translocation, and the transcription of sulfiredoxin. Lack of the active form of Prx-2 leads to a maintained state of oxidative stress and vulnerability to depression. Restoring Nrf2 translocation with 7,8-DHF restores redox homeostasis and transforms vulnerable into NV animals. BDNF, brain-derived neurotrophic factor; CMS, chronic mild stress; DHF, dihydroxyflavone; NV, non-vulnerable; SD, social defeat.
We demonstrate that Nrf2 is a key regulator of oxidative stress following SD. Dysfunction in the Nrf2/Keap1 pathway produces redox imbalance in neurodegenerative, cardiovascular, and pulmonary diseases as well as chronic inflammation.19, 23 Until now, the involvement of Nrf2 in stress-related disorders had not been described. Here, we show that the downregulation of nuclear Nrf2 caused a decrease in its transcriptional activity, as indicated by decreased sulfiredoxin mRNA levels. Because sulfiredoxin catalyzes the recycling of overoxidized-Prx-2, the downregulation of sulfiredoxin caused by the decrease in nuclear Nrf2 explains the significant accumulation of the overoxidized/inactive form of Prx-2 and the permanent state of oxidative stress. The decreased translocation of Nrf2 thus prevents the efficient activation of antioxidant defense mechanisms, which favors redox imbalance. Conversely, recovery in NV animals is made possible by an increase in Nrf2 translocation into the nucleus and the subsequently increased sulfiredoxin levels, which, in turn, increases the Prx-2-dependent detoxification of H2O2 produced by SD.
In examining the upstream mechanisms that control Nrf2, we made the unexpected discovery that BDNF can act as a homeostatic regulator of Nrf2 activity by increasing its nuclear translocation. Despite intense scrutiny in cancer research studies, the precise mechanisms that control Nrf2 translocation remain poorly understood.54 It is therefore difficult to propose a mechanism by which BDNF controls the Nrf2 pathway. Nevertheless, we have found that BDNF tonically induces Nrf2 translocation into the nucleus under basal conditions, thereby constantly activating antioxidant mechanisms. Because numerous enzymatic reactions produce ROS, which must then be cleared, we propose that the BDNF-Nrf2 pathway is part of the normal detoxification processes that are continuously activated in cells. During the acute phase (D5), all animals displayed pro-oxidant conditions due to a BDNF-dependent failure to activate Nrf2-dependent antioxidant defenses. However, we cannot exclude the possibility that other transcription factors were involved in this mechanism of redox homeostasis. At D11, BDNF levels were restored in NV animals, and the Nrf2 system showed signs of hyperactivity while oxidative balance was restored (Figure 5). The lower levels of BDNF in the V animals prevented Nrf2 from restoring normal redox conditions (Figure 5). Our results suggest that the resolution of the initial phase of oxidative stress is an active process that involves multiple steps, including the activity of a regulator molecule, BDNF, and a transcription factor, Nrf2, to control the expression of antioxidant enzymes.
Anatomical alterations of the hippocampus are a hallmark not only of animal models of depression and psychosocial stress55, 56, 57 but also of animals that are vulnerable to depression.5 Here, we focused on the CA3 area. Since oxidative stress extends to other hippocampal subfields, anatomical alterations may also affect other hippocampal cells. In the present study, we show that failure of Nrf2 to translocate to the nucleus is causally linked to anatomical alterations. Nrf2-null mice displayed endogenous reduction of apical dendritic length as V rats.5 Similar Nrf2-dependent mechanisms may be at play in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis, which are characterized by oxidative stress and anatomical alterations.58, 59 Using a proteomic approach, we identified abnormal expression patterns of proteins known to be involved in cell morphology, cell projection/organization, and the regulation of protein ser/thr kinase activity. Most of these proteins, including DPYL2, RhoGDI, CapZ beta, PP2A-alpha, UCHL1, HSC71 and enolase, are prone to oxidative modifications.60, 61, 62, 63 If anatomical alterations clearly indicate damage caused by oxidative stress in neuronal circuits, they are not necessarily causally linked to behavioral alterations, although they may contribute to them.64 Although oxidative stress are mostly in neurons, there were also indications of oxidative stress in some glial cells in V animals. We also observed more activated microglia and phagocytic microglial in the CA3 of V rats, as compared with NV (Supplementary Figure S2c). The observed oxidative damage are more prominent in neurons than glial cells may be due to the latter ability to better compensate and repair damage than neurons. This could be explained by the abundance of reduced glutathione in glial cells than in neurons.65 We cannot rule out a contribution of these cells to the vulnerability phenotype.
Multiple studies have shown that Nrf2 is involved in numerous pathological processes. Activating the Nrf2-Keap1 pathway with chemical activators is beneficial prior to/after intracerebral hemorrhage or traumatic brain injury as well as in chemical/acute models of neurodegeneration.18, 19, 66 Here, we demonstrate that Nrf2 dysfunction is also central to vulnerability to depression in a double-hit scenario. Nrf2-null mice were endogenously in a vulnerable state, as indicated by results showing that mild stress was sufficient to induce a depressive phenotype. Restoring Nrf2 function with an activator of Nrf2 (t-BHQ) or 7,8-DHF, regardless of its mode of action, transformed V into NV animals, as reflected in their abolished vulnerability to depression. Hence, permanent Nrf2 dysfunction is both necessary and sufficient to induce a permanent state of vulnerability to depression (although the necessary and sufficient conditions were tested in different species: rats and mice, respectively). Because depression is associated with most neurological disorders,13 it will be particularly important to assess Nrf2 function in these pathologies. In particular, we recently demonstrated that in a stress-epilepsy double-hit scenario, only vulnerable animals developed depression-like phenotype after the onset of epilepsy, suggesting that a context including high allostatic load may favor the development of depression in neurological disorders.35
Our data show that BDNF is involved in vulnerability to depression rather than in depression per se, which may explain why clinical data do not always support a direct correlation between BDNF levels and depression.67 Since early-life adversity leaves lasting epigenetic marks at the BDNF gene,68 and since the stress disorders involve the epigenetic regulation of Rac1 following SD,64 epigenetic mechanisms may be involved in BDNF/Nrf2-dependent vulnerability depression, at least in the model used here.
In conclusion, our study paves the way for the identification of at-risk patients and preventive treatments. We have proposed that BDNF can be used as a biomarker for vulnerability to depression and that reinstating redox balance restores a normal phenotype. More generally, the BDNF-Nrf2 pathway constitutes an interesting therapeutic target (in particular with t-BHQ) for the treatment of stress-related psychiatric disorders and neurodegenerative diseases that are associated with pro-oxidant conditions.
References
McEwen BS . The brain on stress: toward an integrative approach to brain, body and behavior. Perspect Psychol Sci 2013; 8: 673–675.
McEwen BS . Biomarkers for assessing population and individual health and disease related to stress and adaptation. Metabolism 2015; 64: S2–S10.
de Kloet ER, Joels M, Holsboer F . Stress and the brain: from adaptation to disease. Nat Rev Neurosci 2005; 6: 463–475.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 2007; 131: 391–404.
Blugeot A, Rivat C, Bouvier E, Molet J, Mouchard A, Zeau B et al. Vulnerability to depression: from brain neuroplasticity to identification of biomarkers. J Neurosci 2011; 31: 12889–12899.
Miczek KA, Nikulina EM, Takahashi A, Covington HE 3rd, Yap JJ, Boyson CO et al. Gene expression in aminergic and peptidergic cells during aggression and defeat: relevance to violence, depression and drug abuse. Behav Genet 2011; 41: 787–802.
Terracciano A, Lobina M, Piras MG, Mulas A, Cannas A, Meirelles O et al. Neuroticism, depressive symptoms, and serum BDNF. Psychosom Med 2011; 73: 638–642.
Lang UE, Hellweg R, Gallinat J . BDNF serum concentrations in healthy volunteers are associated with depression-related personality traits. Neuropsychopharmacology 2004; 29: 795–798.
Minelli A, Zanardini R, Bonvicini C, Sartori R, Pedrini L, Gennarelli M et al. BDNF serum levels, but not BDNF Val66Met genotype, are correlated with personality traits in healthy subjects. Eur Arch Psychiatry Clin Neurosci 2011; 261: 323–329.
Avila-Costa MR, Colin-Barenque L, Fortoul TI, Machado-Salas JP, Espinosa-Villanueva J, Rugerio-Vargas C et al. Motor impairments in an oxidative stress model and its correlation with cytological changes on rat striatum and prefrontal cortex. Int J Neurosci 2001; 108: 193–200.
Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M . Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol 2009; 19: 220–230.
Hovatta I, Juhila J, Donner J . Oxidative stress in anxiety and comorbid disorders. Neurosci Res 2010; 68: 261–275.
Maes M, Kubera M, Obuchowiczwa E, Goehler L, Brzeszcz J . Depression's multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Neuro Endocrinol Lett 2011; 32: 7–24.
Bergamini CM, Gambetti S, Dondi A, Cervellati C . Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 2004; 10: 1611–1626.
Mates JM . Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000; 153: 83–104.
Rhee SG . Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006; 312: 1882–1883.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13: 76–86.
Baird L, Dinkova-Kostova AT . The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol 2011; 85: 241–272.
Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P . Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res 2010; 44: 1267–1288.
Soriano FX, Baxter P, Murray LM, Sporn MB, Gillingwater TH, Hardingham GE . Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol Cells 2009; 27: 279–282.
Biteau B, Labarre J, Toledano MB . ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003; 425: 980–984.
Abbas K, Breton J, Planson AG, Bouton C, Bignon J, Seguin C et al. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radic Biol Med 2011; 51: 107–114.
Halliwell B . Oxidative stress and neurodegeneration: where are we now? J Neurochem 2006; 97: 1634–1658.
Maes M, Fisar Z, Medina M, Scapagnini G, Nowak G, Berk M . New drug targets in depression: inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates—Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 2012; 20: 127–150.
Martín-de-Saavedra MD, Budni J, Cunha MP, Gómez-Rangel V, Lorrio S, Del Barrio L et al. Nrf2 participates in depressive disorders through an anti-inflammatory mechanism. Psychoneuroendocrinology 2013; 38: 2010–2022.
de Boer SF, van der Vegt BJ, Koolhaas JM . Individual variation in aggression of feral rodent strains: a standard for the genetics of aggression and violence? Behav Genet 2003; 33: 485–501.
Andre J, Zeau B, Pohl M, Cesselin F, Benoliel JJ, Becker C . Involvement of cholecystokininergic systems in anxiety-induced hyperalgesia in male rats: behavioral and biochemical studies. J Neurosci 2005; 25: 7896–7904.
Becker C, Thiebot MH, Touitou Y, Hamon M, Cesselin F, Benoliel JJ . Enhanced cortical extracellular levels of cholecystokinin-like material in a model of anticipation of social defeat in the rat. J Neurosci 2001; 21: 262–269.
Kasai H . Analysis of a form of oxidative DNA damage, 8-hydroxy-2'-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res 1997; 387: 147–163.
Shih AY, Li P, Murphy TH . A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci 2005; 25: 10321–10335.
Benoliel JJ, Mauborgne A, Bourgoin S, Legrand JC, Hamon M, Cesselin F . Opioid control of the in vitro release of cholecystokinin-like material from the rat substantia nigra. J Neurochem 1992; 58: 916–922.
Wilcox CS, Pearlman A . Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev 2008; 60: 418–469.
Wilcox CS . Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol Ther 2010; 126: 119–145.
Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA 2010; 107: 2687–2692.
Becker C, Bouvier E, Ghestem A, Siyoucef S, Claverie D, Camus F et al. Predicting and treating stress-induced vulnerability to epilepsy and depression. Ann Neurol 2015; 78: 128–136.
Flint DH, Tuminello JF, Emptage MH . The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem 1993; 268: 22369–22376.
Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM et al. GO:TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004; 20: 3710–3715.
Wassmann S, Wassmann K, Nickenig G . Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 2004; 44: 381–386.
Cocheme HM, Logan A, Prime TA, Abakumova I, Quin C, McQuaker SJ et al. Using the mitochondria-targeted ratiometric mass spectrometry probe MitoB to measure H2O2 in living Drosophila. Nat Protoc 2012; 7: 946–958.
Rothman SM, Mattson MP . Adverse stress, hippocampal networks, and Alzheimer's disease. Neuromolecular Med 2010; 12: 56–70.
Bell KF, Hardingham GE . CNS peroxiredoxins and their regulation in health and disease. Antioxid Redox Signal 2011; 14: 1467–1477.
Sartorius A, Hellweg R, Litzke J, Vogt M, Dormann C, Vollmayr B et al. Correlations and discrepancies between serum and brain tissue levels of neurotrophins after electroconvulsive treatment in rats. Pharmacopsychiatry 2009; 42: 270–276.
Schmidt HD, Duman RS . Peripheral BDNF produces antidepressant-likeeffects in cellular and behavioral models. Neuropsychopharmacology 2010; 35: 2378–2391.
Zhang R, Kang KA, Piao MJ, Ko DO, Wang ZH, Chang WY et al. Preventive effect of 7,8-dihydroxyflavone against oxidative stress induced genotoxicity. Biol Pharm Bull 2009; 32: 166–171.
Chen J, Chua KW, Chua CC, Yu H, Pei A, Chua BH et al. Antioxidant activity of 7,8-dihydroxyflavone provides neuroprotection against glutamate-induced toxicity. Neurosci Lett 2011; 499: 181–185.
Gupta VK, You Y, Li JC, Klistorner A, Graham SL . Protective effects of 7,8-dihydroxyflavone on retinal ganglion and RGC-5 cells against excitotoxic and oxidative stress. J Mol Neurosci 2013; 49: 96–104.
Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A et al. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol 2006; 175: 779–789.
Rhee SG, Woo HA, Kil IS, Bae SH . Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem 2012; 287: 4403–4410.
Przedborski S . Peroxiredoxin-2 links Cdk5 to neurodegeneration. Nat Med 2007; 13: 907–909.
Conrad CD . What is the functional significance of chronic stress-induced CA3 dendritic retraction within the hippocampus? Behav Cogn Neurosci Rev 2006; 5: 41–60.
McEwen BS . Stress, sex, and neural adaptation to a changing environment: mechanisms of neuronal remodeling. Ann NY Acad Sci 2010; 1204: E38–E59.
Serhan CN . The resolution of inflammation: the devil in the flask and in the details. FASEB J 2011; 25: 1441–1448.
Troncoso Brindeiro CM, Lane PH, Carmines PK . Tempol prevents altered K(+) channel regulation of afferent arteriolar tone in diabetic rat kidney. Hypertension 2012; 59: 657–664.
Xiang M, Namani A, Wu S, Wang X . Nrf2: bane or blessing in cancer? J Cancer Res Clin Oncol 2014; 140: 1251–1259.
Becker C, Zeau B, Rivat C, Blugeot A, Hamon M, Benoliel JJ . Repeated social defeat-induced depression-like behavioral and biological alterations in rats: involvement of cholecystokinin. Mol Psychiatry 2008; 13: 1079–1092.
Krishnan V, Nestler EJ . The molecular neurobiology of depression. Nature 2008; 455: 894–902.
Ota KT, Duman RS . Environmental and pharmacological modulations of cellular plasticity: role in the pathophysiology and treatment of depression. Neurobiol Dis 2013; 57: 28–37.
Carri MT, Ferri A, Cozzolino M, Calabrese L, Rotilio G . Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull 2003; 61: 365–374.
Jenner P . Oxidative stress in Parkinson's disease. Ann Neurol 2003; 53: S26–S36, discussion S-8.
Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R et al. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem 2002; 82: 1524–1532.
Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D . Differential proteome analysis of aging in rat skeletal muscle. FASEB J 2005; 19: 1143–1145.
Ciavardelli D, Silvestri E, Del Viscovo A, Bomba M, De Gregorio D, Moreno M et al. Alterations of brain and cerebellar proteomes linked to Abeta and tau pathology in a female triple-transgenic murine model of Alzheimer's disease. Cell Death Dis 2010; 1: e90.
Foley TD, Melideo SL, Healey AE, Lucas EJ, Koval JA . Phenylarsine oxide binding reveals redox-active and potential regulatory vicinal thiols on the catalytic subunit of protein phosphatase 2A. Neurochem Res 2011; 36: 232–240.
Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med 2013; 19: 337–344.
Raps SP, Lai JC, Hertz L, Cooper AJ . Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res 1989; 493: 398–401.
van Muiswinkel FL, Kuiperij HB . The Nrf2-ARE Signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord 2005; 4: 267–281.
Fernandes BS, Gama CS, Cereser KM, Yatham LN, Fries GR, Colpo G et al. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res 2011; 45: 995–1004.
Roth TL, Lubin FD, Funk AJ, Sweatt JD . Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry 2009; 65: 760–769.
Acknowledgements
This research was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM), Université Pierre et Marie Curie and fellowships from Medicen and for National Center of Competence in Research 'SYNAPSY' 51AU40_125759 (to KQD); Avina Foundation (to KQD). Wild-type Groningen strain was generously provided by De Boer S. (University of Groningen, The Netherlands). We thank Dr Henri Gozlan, Françoise Camus, Amandine Mouchard, Céline Drieu, Evelyne Donsez, Annabelle Reaux-Le-Goazigo and Pr Michel Cuenod for their valuable advice and helpful comments. We thank S Riquier, D Tondelier and A Edelman for 2D gel electrophoresis and mass spectrometry analysis, JC Drapier for helpful discussions on redox homeostasis and signaling. We thank Dr B Giros for critical reading of the manuscript. We thank V Vialou for the SD in mice.
Author Contributions
EB performed experiments, interpreted data; FB performed experiments, interpreted redox data, designed research and contributed to writing of the manuscript; JM performed experiments and analyzed; J-HC performed immunohistochemistry; DC performed RT–PCR experiments; NC performed experiments; ND performed experiments; CR interpreted experiments; KQD designed research; CB designed research and contributed to writing of the manuscript; JJB designed research, analyzed data, interpreted data, contributed to writing of the manuscript and supervised the project. CB advised, designed research, performed experiments, analyzed data and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Bouvier, E., Brouillard, F., Molet, J. et al. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol Psychiatry 22, 1701–1713 (2017). https://doi.org/10.1038/mp.2016.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.144
This article is cited by
-
Focusing on mitochondria in the brain: from biology to therapeutics
Translational Neurodegeneration (2024)
-
Sex-specific interactions between stress axis and redox balance are associated with internalizing symptoms and brain white matter microstructure in adolescents
Translational Psychiatry (2024)
-
Ferroptosis regulation through Nrf2 and implications for neurodegenerative diseases
Archives of Toxicology (2024)
-
Nrf2 regulates iron-dependent hippocampal synapses and functional connectivity damage in depression
Journal of Neuroinflammation (2023)
-
Self-immolative nanocapsules precisely regulate depressive neuronal microenvironment for synergistic antidepression therapy
Journal of Nanobiotechnology (2023)
