
Abstract
Variants in CLCN4, which encodes the chloride/hydrogen ion exchanger CIC-4 prominently expressed in brain, were recently described to cause X-linked intellectual disability and epilepsy. We present detailed phenotypic information on 52 individuals from 16 families with CLCN4-related disorder: 5 affected females and 2 affected males with a de novo variant in CLCN4 (6 individuals previously unreported) and 27 affected males, 3 affected females and 15 asymptomatic female carriers from 9 families with inherited CLCN4 variants (4 families previously unreported). Intellectual disability ranged from borderline to profound. Behavioral and psychiatric disorders were common in both child- and adulthood, and included autistic features, mood disorders, obsessive–compulsive behaviors and hetero- and autoaggression. Epilepsy was common, with severity ranging from epileptic encephalopathy to well-controlled seizures. Several affected individuals showed white matter changes on cerebral neuroimaging and progressive neurological symptoms, including movement disorders and spasticity. Heterozygous females can be as severely affected as males. The variability of symptoms in females is not correlated with the X inactivation pattern studied in their blood. The mutation spectrum includes frameshift, missense and splice site variants and one single-exon deletion. All missense variants were predicted to affect CLCN4’s function based on in silico tools and either segregated with the phenotype in the family or were de novo. Pathogenicity of all previously unreported missense variants was further supported by electrophysiological studies in Xenopus laevis oocytes. We compare CLCN4-related disorder with conditions related to dysfunction of other members of the CLC family.
Similar content being viewed by others


Introduction
Chloride is the most abundant anion in vivo and is actively transported and its level tightly regulated in virtually all animal cells.1 Mammals express nine chloride channel (CLC) genes, five of which encode 2Cl−/H+ exchangers (CIC-3-7) and four Cl− channels (CIC-1,-2,-Ka,-Kb).2 The mainly endosomal/lysosomal 2Cl−/H+ exchangers may provide an electric shunt to facilitate vesicular acidification and accumulate vesicular chloride. In contrast, CLC Cl− channels mainly reside in the plasma membrane and are involved in the regulation of electrical excitability or control extra- and intracellular ion homeostasis.2 The physiological relevance of all members of the CLC gene family is highlighted by mutations in various human diseases and mouse models, with phenotypes including leukodystrophy, lysosomal storage disease, severe neurodegeneration, myotonia, deafness, male infertility, impaired renal function and osteopetrosis.2
Inherited single-nucleotide variants in CLCN4 (MIM *302910), which encodes the 2Cl−/H+ exchanger CIC-4,3, 4 were recently identified in five families with variable X-linked intellectual disability (ID) and seizure disorder,5 one de novo variant was identified in a male with epileptic encephalopathy6 and one de novo variant was reported in a female with borderline ID and congenital diaphragmatic hernia.7 In humans, CIC-4 is widely expressed across tissues with particularly high expression in the brain.8 ClC-4 resides predominantly on intracellular vesicles, but remains in the endoplasmic reticulum, with a minor portion reaching the plasma membrane when overexpressed heterologously.9 Its physiological function is unclear, but CIC-4 is likely to be involved in the ion homeostasis of endosomes and intracellular trafficking.2 A role for CIC-4 in neuronal differentiation has been suggested based on studies using primary hippocampal neurons from Clcn4−/− mice and wild-type neurons subjected to short hairpin RNA-mediated Clcn4 knockdown, which showed a modest reduction in the total number of dendritic branches per neuron.5 However, the pathophysiology of CLCN4-related disorder in humans is not yet understood.
We report 10 additional families with nine novel CLCN4 variants, extend the molecular spectrum to include splice site variants and single-exon deletions, suggest genotype–phenotype correlation, and present detailed clinical phenotypic information about CLCN4-related disorder in 29 hemizygous males and 23 heterozygous females from 16 families. Pathogenicity of previously unreported missense variants is supported by electrophysiological studies in Xenopus laevis oocytes.
Materials and methods
Families A, C, D, E and F were part of an X-chromosome exome resequencing project reported by Hu et al.,5 with limited clinical information provided. Family A was originally described by Claes et al.,10 as MRX49 (MIM: 300114) (Figure 1); all available affected individuals (A:III:6, A:IV:2 and A:IV:4) were reviewed by HVE (aged 52, 27 and 33) in 2015. Family C was originally described by Raynaud et al.,11 as MRX15 (MIM: 300115) (Figure 1) and was unavailable for review in 2016. The variant for the proband in family K was reported by Yu et al.,7 as part of a case series of patients with congenital diaphragmatic hernia but detailed clinical information was not provided. Ten families (B, G–J and L–P) are previously unreported. Genetic studies were approved by local ethics committees. Written informed consent was obtained for molecular genetic analysis and the publication of clinical and radiological data and photographs from all participants or their legal guardians.

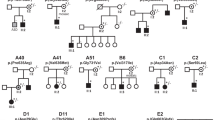
Pedigrees of affected families A–G, J and P. Filled square=affected male; Filled circle=affected female; *Familial CLCN4 variant present; wt, familial CLCN4 variant absent; NT, not tested. Note unaffected male IV:5 in family B has Klinefelter syndrome (47,XXY). Probands from families H, I, K, L, M, N and O (pedigrees not shown) have de novo variants.
Probands from families A, C-F, G, I and K-P had next-generation sequencing (X-exome or whole-exome sequencing: details of laboratories and methodologies are given in Supplementary Table 2);5, 7, 12,13,14 the proband from family B was investigated by droplet-based multiplex PCR (7367 amplicons, 757 X-chromosome genes, 1.54 Mb),5, 15 the proband from family G had targeted sequencing of the coding exons of X-chromosomal genes as part of a large-scale resequencing screen.16 Tested members of family J had a clinical chromosomal microarray (v10.2 Baylor Miraca Genetics Laboratory, Houston, TX, USA). The pathogenicity of missense variants was assessed using in silico tools including combined annotation-dependent depletion score17 and PolyPhen. Confirmation of variants and segregation analysis was conducted by Sanger sequencing using standard methods.
Analogous positions of amino acids mutated in CIC-4 were modeled in the three-dimensional crystal structure of the algal CmClC, as previously described.5 Missense variants were introduced into mammalian and oocyte expression vectors by recombinant PCR in order to study their functional effects. The subcellular localization of mutant CIC-4 proteins was investigated upon overexpression in COS-7 cells. CLCN4 variants were also expressed in Xenopus laevis oocytes and their effect on the outward rectifying CIC-4-mediated currents was measured as previously described.5
Results
Pedigrees for families with inherited variants are provided in Figure 1, with clinical information summarized in Table 1 and additional clinical information for each family provided in Supplementary Table 1.
Global developmental delay and ID was reported for all males (n=29) and variable even within families, with severity varying from borderline to severe. Expressive and receptive language was commonly an area of particular concern. Ten (34%) males were reported to be of amiable personality with no significant challenging behaviors or mental health concerns. Significant behavioral or mental health issues were noted in 19 (66%) males: hetero-aggressive behavior was reported in 8 males, auto-aggressive behavior in 3 males, repetitive autistic or obsessive–compulsive like behaviors in 7 males and hyperactivity in 3 males. Depressive or anxiety symptoms were reported in seven males (24%), including two who had a diagnosis of bipolar disorder. A seizure disorder was reported in 15 males (52%) from seven families varying from infrequent seizures, well controlled with monotherapy (47%), to severe infantile-onset intractable epilepsy (53%), even within a family. Seizure types included infantile spasms, absence, myoclonic, tonic, complex partial and generalized tonic-clonic. Three seizure-related deaths were reported in family F, which may have resulted from limited anti-epileptic treatment options. Reported EEG findings included focal spikes, multifocal spikes and generalized background slowing (data not shown). Infantile hypotonia was common: reported in eight (27%) males across six families. Additional neurological signs in childhood included strabismus (3), cortical visual impairment (2), gait abnormality (1), upper limb choreiform movements (1) and evolving upper limb/generalized spasticity in childhood (2). Progressive ataxia and/or lower limb spasticity were described after the third decade in four males from two families (F and G). Abnormalities on neuroimaging/neuropathology were noted in seven males across five families (64% of tested individuals, Figure 2). However, the majority of males did not have neuroimaging and brain magnetic resonance imaging (MRI) was normal in an additional four males. White matter abnormalities and cortical atrophy were characteristic: periventricular leukomalacia and/or ventricular dilatation was noted in childhood in three males with additional features of delayed myelination and hypoplastic corpus callosum (for example, Figure 2:J1–6); one male had cortical atrophy noted in his 20s and another male had marked supra- and infra-tentorial atrophy and callosal thinning in his 60s (Figure 2:F1–4). A hypoplastic corpus callosum was noted on autopsy of a male who died aged 19 of a seizure-related death.

Neuroimaging of affected individuals with CLCN4-related condition at different ages. F1–4: CT images of individual F:IV:3, aged 66. Non-contrast multi-slice helical acquisitions, thick-slice reconstructions only. F1: sagittal midline image depicts callosal thinning, atrophic midbrain and cerebellar atrophy with prominent quadri-cerebellar cistern. F2: axial image of posterior fossa depicting cerebellar vermian atrophy with increased caliber of the adjacent CSF spaces. F3: axial image depicting prominent Sylvian fissures and increased caliber of lateral and third ventricles with surrounding atrophic change and evidence of a right frontal infarct. F4: coronal image showing disproportionate cerebellar atrophy and prominence of the adjacent CSF spaces. J1–6: MRI images of individual J:III:2 aged 4 and 8. J1: Axial FLAIR image from individual J:III:2, aged 4, depicting enlarged Sylvian fissures secondary to surrounding volume loss, particularly affecting the frontal lobes. J2: coronal T2 weighted image from individual J:III:2 aged 4 depicting generalized white matter volume loss. J3 Axial FLAIR image from individual J:III:2 aged 8 demonstrating periventricular leukomalacia. J4: coronal FLAIR image from individual J:III:2 aged 8. Images depict that despite interval progression of myelination between the ages of 4 and 8, there is still significantly delayed myelination involving the anterior frontal, posterior parietal and temporal lobes. The corpus callosum is normally formed but attenuated. There is pronounced global volume loss, preferentially affecting the white matter. Prominent perivascular spaces are seen bilaterally. L1–3: MRI images from family L proband at four months of age. Diffuse cortical hypoplasia with resultant prominence of the extra-axial CSF spaces and thinning of the corpus callosum without other morphologic abnormalities. Myelination is otherwise grossly normal. N1–2 MRI images from proband from family N aged 10. N1: sagittal T1 weighted midline image showing dilated third ventricle. N2: Coronal T2 weighted image showing dilated third ventricle. I1–2: images from proband from family I aged 13 months. I1: sagittal T1 weighted midline image showing dilated lateral ventricles. I:2: axial T2 weighted image showing volume loss, periventricular leukomalacia and dilated ventricles. CSF, cerebrospinal fluid; MRI, magnetic resonance imaging.
Although the previously reported adult members of families A (MRX49)10 and C (MRX15)11 were considered to be non-dysmorphic, when reassessed with affected members of families B and F (Figure 3) and D and G (TK and MF review), common dysmorphic features in older male individuals included a long face with straight nose and a prominent pointed chin that becomes more ‘squared off’ with age, and a relatively flat midface. Facial features were not so characteristic in younger males. Growth parameters were generally within the normal range; however, many older individuals had a lean body habitus; some males had macrocephaly (families A, F and P), whereas microcephaly was noted in males from families B, I and O. No consistent pattern of extra-neurocognitive symptoms was reported, and many noted features are commonly seen in children with ID of other causes: two affected infants had gastro-esophageal reflux and aspiration/apnoeic events in early infancy, and feeding difficulties were significant for probands from families I and O. Three affected males in family C had scoliosis and the affected male in family P had a unilateral inguinal hernia, cryptorchidism and pes planus.

Facial features of affected individuals with CLCN4-related disorder in age order. In older males note the elongated face, straight nose and pointed prominent chin that becomes relatively ‘squared off’ with age that is particularly prominent in affected adult males from families A, B, C and F (F:IV:3), and mildly present in the 9-year-old F:V:1 and 12-year-old affected male from family P. A relatively flat midface is noticeable in several individuals (particularly A:III:6 and affected members of family C). Individuals F:IV:3 and F:IV:5 have relative ‘coarsening’ of facial features with age; however, they were both treated with phenytoin, which can coarsen features and F:IV:5 had multiple facial injuries secondary to epileptic drop attacks. Although the facial phenotype is less distinctive in younger children and females (family F:V:1 and F:V2 as infants, and affected individuals from families O, I, L, H, N and M) some similarities include young children having relatively rounded faces and affected females from families H, L and F (F:IV:5) having down-sloping palpebral fissures and depressed nasal bridges. Clinical pictures from family C were first published in Raynaud et al.11 and reproduced from John Wiley & Sons with permission.
The phenotype in females (n=18) from families with inherited CLCN4 variants was strikingly variable, ranging from unaffected to severely affected, even within a family (for example, family F). No phenotype was recognized in tested/obligate heterozygous females from families B, C, E, J and P. Two obligate female heterozygotes from family A were noted to have subtle learning/behavioral difficulties. Three heterozygous females from family F were asymptomatic, one had neuropsychiatric features and one had severe ID with intractable seizure disorder, cerebral atrophy and progressive spasticity and ataxia.
The phenotype in the five heterozygous females with de novo variants was more severe and consistent with the phenotype in hemizygous males. One had borderline, two had moderate and two had severe-profound ID; again language development, especially expressive language, was an area of particular concern. Two girls (40%) had seizure disorders of varying severity. Two girls had self-injurious behaviors and one was assessed as emotionally reactive; the other two girls had no significant behavioral issues. Three girls (60%) had infantile hypotonia and one developed mildly increased peripheral tone with brisk patellar reflexes. Four girls had neuroimaging by MRI: reported as normal for two (both assessed below the age of three). There was subtle neuropathology in the other two with diffuse cortical and corpus callosum hypoplasia in one, and persistently enlarged third ventricles and T2/FLAIR subcortical hyperintensities in the other (Figure 2). Two of the five girls had significant feeding difficulties in infancy; one had congenital diaphragmatic hernia and bilateral hip dysplasia, and one had scoliosis. Two of the girls had microcephaly: other growth parameters were within normal limits (Supplementary Table 1). Subtle dysmorphic features were noted in three girls (Figure 3) including down-sloping palpebral fissures with depressed nasal bridge. We are also aware (JR) of another female (not included in this study) with a novel de novo missense variant in CLCN4, p.Ile195Phe, with a similar clinical phenotype including ID, infantile hypotonia, autistic features, focal seizures, microcephaly and periventricular leukomalacia/hypomyelination on cerebral MRI.
X inactivation studies were performed for eight female heterozygotes. In an asymptomatic heterozygote from family F, X inactivation was skewed (87%:13%); in all other females it was random or non-informative (Supplementary Information). None of the severely affected females had an alternative cause identified for their clinical features when subjected to other genetic (including chromosomal microarray) and metabolic testing, including whole-genome sequencing for the severely affected female in family F.
In addition to the one frameshift variant (p.Asp15Serfs*18, family A) and four missense variants (p.Gly731Arg, p.Gly78Ser, p.Leu221Val, p.Val536Met; families C-F) reported in Hu et al.5 and the p.Asp15Asn in family K reported by Yu et al.,7 an additional novel CLCN4 frameshift variant was identified in family B (p.Ile626Asnfs*135), six novel missense CLCN4 variants were detected in families G, H, I, L, M and N (p.Val212Gly, p.Leu221Pro, p.Ser534Leu, p.Ala555Val, p.Arg718Trp and p.Val275Met), and a novel intragenic microdeletion of exon 12 (hg19 chrX: g. (10182428_10187807)_(10189796_10201480) del mat) predicted to result in a frameshift and premature stop codon was detected in family J (Supplementary Table 2, Figure 4, Supplementary Figure 3). The proband in family O had the same p.Gly544Arg missense variant (but a different nucleotide substitution) as the affected individual with epileptic encephalopathy reported by Veeramah et al.6 In proband O, this missense variant appeared to be de novo but present in mosaic form (70% of the cells). The proband and his unaffected mother in family P had a novel intronic variant (IVS9+5G>A) predicted to affect splicing leading to in-frame deletion of exon 9. Exon 9 would be predicted to be a critical exon as it codes for four helical transmembrane domains.

Genotypic information, three-dimensional modeling and functional studies of CLCN4 variants. (a) Position of CLCN4 variants in families A–P (information adapted from Protein Data Bank P51793). Missense variants in families D (p.Gly78Ser), G (p.Val212Gly), L (p.Ala555Val) and O (p.Gly544Arg) are within helical transmembrane domains of the protein. Missense variants in families F (p.Val536Met), I (p.Ser534Leu) and N (p.Val275Met) are within helical-intramembrane domains. Missense variants in families E (p.Leu221Val) and H (p.Leu221Pro) lie within a loop between transmembrane domains. Missense variants in families C (p.Gly731Arg) and M (p.Arg718Trp) lie within the second cytosolic cystathionine-β-synthase (CBS) domain and therefore may affect gating. (b) Analogous positions of amino acids mutated in ClC-4 highlighted in the crystal structure of CmClC. Amino acids are displayed as spheres in colors. CLC transporters are (homo)dimers of identical subunits with two ion translocation pathways. Each subunit is composed of a transmembrane domain (TMD) containing several intramembrane helices and cytosolic CBS domains. Asterisks indicate previously published analogous positions of amino acids.5, 6 (c) Current voltage relationships of the electrogenic 2Cl−/H+ exchanger ClC-4 and the CIC-4 variants expressed in Xenopus oocytes, shown as mean values of normalized steady-state currents from several oocytes (numbers indicated in figure, in parentheses: number of independent cRNA batches injected). The current of wild-type (WT) CIC-4 is strongly outwardly rectifying, that is, increases steeply at inside-positive voltages. The human missense mutations p.Val212Gly, p.Leu221Pro, p.Val275Met, p.Ser543Leu, p.Ala555Val and p.Arg718Trp reduced, or even abolished, these ClC-4 currents. Only variant p.Asp15Asn exhibited normal wild-type like currents. Error bars, s.e.m. Noninjected (n.i.) oocytes served as control.
To test the in silico prediction, for variant IVS9+5G>A, we performed reverse transcriptase-PCR experiments on RNA extracted from lymphoblastoid cell lines using CLCN4 primers located in exon 8 and exon 10. In the proband and his mother, two specific amplification products were obtained, with the larger product corresponding to properly spliced wild-type CLCN4 transcripts with exon 9 spliced in and the smaller product corresponding to transcripts with exon 9 skipped (Supplementary Figure 2). In contrast, a single wild-type product was seen in controls (Supplementary Figure 2). Skipping of exon 9 would result in mutant ClC-4 protein with 182 amino acids deleted, supportive of the in silico pathogenicity prediction. The results also indicated that the mutated allele is not X-inactivated in the unaffected mother’s blood lymphocytes.
The accession numbers for the novel sequences reported in this paper are ClinVar: SCV000245780–SCV000245788, SCV000266304 and SCV000266305.
All missense variants were predicted to affect function by in silico software analysis, were not listed in publicly available databases including ExAC, affected highly evolutionary conserved amino acids (Supplementary Figure 1) and either segregated with the condition in the family (Figure 1) or were de novo, with no other plausible genetic cause detected on exome sequencing and chromosomal microarray (Supplementary Information).
We examined the position of the novel missense variants using the RCSB protein bank (Figure 4a) and modeled the analogous positions of amino acids mutated in CIC-4 in the three-dimensional crystal structure of CmCLC, a Cl−/H+ antiporter from Cyanidioschyzon merolae18 (Figure 4b and Supplementary Table 2). The majority of the missense variants affect residues in the transmembrane part that contains the ion translocation pathway, often close to the interface of the two subunits of the dimeric transporter. Two variants are located, also close to the interface, in the cystathionine-β-synthase domains, which have a role in the voltage-dependent activation of CLC channels19 and transporters.20 The previously reported de novo missense variant in family K (p.Asp15Asn)7 is located in the N-terminal cytoplasmic domain, which is not resolved in the crystal (Supplementary Table 2). The exonic deletion present in family J (Supplementary Figure 3A) did not overlap with any predicted benign copy number variants in curated databases (Database of Genomic Variation) and no overlapping variants were present in the laboratory database. This array finding was independently validated in a separate laboratory (Supplementary Figure 3B). The intronic variant in family P was not listed in ExAC and affects a highly conserved nucleotide (Supplementary Figure 1).
These genotypic findings are consistent with, and extend beyond those of Hu et al.5 As previously shown for the ClC-4 variants reported by Hu et al.5 and Veeramah et al.,6 we found that the novel variants p.Val212Gly, p.Leu221Pro, p.Ser534Leu, p.Ala555Val, p.Arg718Trp and p.Val275Met, reduced or abolished the strongly outwardly rectifying CIC-4 currents (Figure 4c). In comparison, the previously reported variant p.Asp15Asn7 in family K yielded wild-type like currents when heterologously expressed (Figure 4c). Western blots of individual oocytes showed equal CIC-4 protein expression levels for all mutants (Supplementary Figure 4). Similar to overexpressed wild-type ClC-4 and the previously reported CLCN4 missense variants,5, 6 all examined missense variants localized predominantly to the endoplasmic reticulum when overexpressed in COS-7 cells (Supplementary Figure 5). However, as native CIC-4 may localize to endosomal compartments,21 this does not exclude a trafficking defect in vivo.
Discussion
This case series indicates that the CLCN4-related condition should be considered a syndromic form of X-chromosome-linked ID, behavioral disorder and epilepsy with complete penetrance but variable expressivity in males, and incomplete penetrance and variable expressivity in females.
Although a larger case series will be required to precisely define phenotype–genotype correlations and for this we are curating a publically accessible CLCN4 website, as part of the Human Disease Genes Website Series, some initial observations can be made. Most variants are missense rather than protein truncating. Three mutations are likely to result in a complete loss of ClC-4 function in hemizygous males; these are frameshift variants in exons 3 and 11 (families A and B) and deletion of exon 12 (family J). Phenotypes of males and females with frameshifts or exonic deletion are relatively mild compared with the majority of affected individuals with missense variants (Supplementary Table 1), which may suggest that an abnormally functioning CIC-4 antiporter is more deleterious than a reduction in its levels or a complete absence. It is tempting to speculate that this stronger effect of missense variants may be mediated by a heteromerization of the mutated ClC-4 protein with homologous CLC proteins.22 However, no potential dominant-negative effect of CLCN4 missense variants was observed when equal amounts of wild-type and mutant ClC-4 were expressed in Xenopus oocytes. The corresponding currents could not be distinguished from a superimposition of currents from WT and mutant ClC-4 transporters (Supplementary Figure 6).
One variant is recurrent: the male in family O has the same amino-acid change (p.Gly544Arg) as previously reported by Veeramah et al.,6 although in mosaic form, and there are phenotypic similarities with both affected males having severe developmental delay/ID, a refractory seizure disorder and microcephaly. There are two families (H and E) where the same amino acid is mutated but to different amino acids. We compared the clinical phenotype and the electrophysiological effect of these variants when heterologously expressed. In family H, the heterozygous female with a de novo alteration of leucine 221 to proline, is affected with moderate developmental delay and hypotonia with no seizure disorder (Supplementary Table 1). In family E, where leucine 221 is altered to valine, affected males have a relatively mild phenotype (mild–moderate ID and only one with a well-controlled seizure disorder) and female heterozygotes are unaffected. The p.Leu221Pro variant has a Grantham score of 98 as opposed to p.Leu221Val, which has a Grantham score of 32.23 We recognize increasing caution in the ion channel literature regarding the accuracy of in silico tools in interpreting clinical severity. For example, although Brunklaus et al.24 found some correlations between predicted physico-chemical differences (using Grantham scores) of >1300 variants in a range of sodium ion channel genes and the clinical severity of the corresponding cardiac, muscular or neurological disorder, they also acknowledged the great variability in clinical expression among members of the same mouse or human family, postulating the importance of differences in genetic background between individuals. Nevertheless, when heterologously expressed in Xenopus oocytes the ClC-4 p.Leu221Pro variant identified in our study essentially abolished the CIC-4 current (Figure 4c) as opposed to the Leu221Val variant, which only resulted in modest reduction in CIC-4 current (Hu et al.5; Figure 1b). Although these genotypic differences may contribute to the differences in clinical phenotypes in females between families E and H, more complex genetic reasons may be contributory. For example, disparities may be attributed to differences in genomic backgrounds and X inactivation patterns in the brain.
The striking phenotypic variability in females with CLCN4-related disorder is similar to that seen in several other X-linked conditions, such as female heterozygotes with mutations in PHF6 (ref. 25) (MIM:300414) and DDX3X (ref. 26) (MIM:300160). Although CLCN4 is subject to X inactivation, blood DNA-based X inactivation studies did not predict clinical outcome in heterozygous females. This is perhaps not surprising as recent mouse studies27 demonstrate the complexity of X inactivation in different cells and organs, so that X inactivation in peripheral blood cells may not reflect X inactivation patterns in the same individual’s brain.
For both affected females and males from the families investigated here, the phenotype is primarily neurocognitive. Only one individual had additional congenital anomalies that is the female in family K (p.Asp15Asn) with a congenital diaphragmatic hernia.7 It is not clear that this anomaly was caused by the CLCN4 variant: for example, no other individuals with CDH described in Yu et al.’s7 case series had variants in CLCN4 and there is no supportive evidence from mouse models that CIC-4 is important in diaphragm development.11 In addition, the pathogenicity of this cytoplasmic variant remains uncertain; although it is de novo and affects an amino acid conserved between species, electrophysiological analysis of Asp15Asn expressing oocytes yielded wild-type currents (Figure 4c). We have listed this variant as a variant of uncertain significance in ClinVar (accession number SCV000266305).
Additional longitudinal clinical data and serial neuroimaging, in particular imaging that is more sensitive to subtle white matter abnormalities, such as FLAIR images (MRI) and PET, will clarify how common white matter changes, cortical atrophy and progressive neurological symptoms are in CLCN4-related disorder, and whether there is a causal association, or whether some of the changes could be acquired. Progressive neurological/neuroanatomical features were noted for individuals with and without a seizure disorder, suggesting that these features were not due solely to uncontrolled seizures causing excitotoxic damage, as has been suggested for other conditions.28 Variable neurological features and leukodystrophy have been described in humans and mice with variants in other members of the CLC protein family.2 Autosomal recessive loss-of-function variants in CLCN2 (MIM *600570) cause a leukoencephalopathy associated with myelin vacuolation in the brain and spinal cord in mice29 and humans.30 Patients with CLCN2 variants display variable neurocognitive symptoms, including ataxia, mild spasticity, visual field defects and learning disabilities.30 Clcn3−/− mice display severe progressive neurodegeneration.31 Clcn6−/− mice have moderate behavioral abnormalities and a mild form of lysosomal storage disease predominantly affecting neuronal axons.32 Neither gene is currently associated with a recognised human phenotype. Lastly, cerebral atrophy and learning difficulties have been described in individuals affected by autosomal recessive CLCN7-related disorder (MIM #611490) and knock-out mice display lysosomal storage/neuronal ceroid lipofuscinosis features.33
More work is required to understand the pathophysiology of CLCN4-related conditions in humans. Decrease in CIC-4 Cl−/H+-exchange activity may impair the ion homeostasis of endosomes and intracellular trafficking.2 No obvious white matter changes or neurodegeneration in the brains of Clcn4−/− mice were observed when examined with luxol staining for myelin (Supplementary Figure S7). However, moderate abnormalities in dendritic branching were reported for cultured hippocampal neurons from Clcn4−/− mice and for wild-type neurons subjected to short hairpin RNA-mediated Clcn4 knockdown,5 changes reminiscent of those seen in other neurodevelopmental conditions.34 The fact that Clcn4−/− mice do not show a recognizable neurological or behavioral phenotype5, 35 could reflect differences in the role of CIC-4 between species or in compensating mechanisms between species. Moreover, Clcn4 is autosomal (on chromosome 7) and has truncated introns in the mouse species Mus musculus, whereas CLCN4 orthologues are on the X-chromosome in other animals and mouse species such as Mus spretus.36 Potentially this could have implications for differential gene expression timing, regulation and subsequent function between CLCN4 and Clcn4 in the humans and mouse. Indeed the rat, where the CLCN4 orthologue is on the X-chromosome, may be an attractive additional animal model for future research.
Currently, treatment for this condition is limited to early intervention, special education, family support and treatment for associated seizure, behavioral, sleep and mental health conditions. The degree of ID was more severe in affected individual(s) from families with a more severe seizure phenotype (for example, families F and J), supporting the prioritization of aggressive seizure control to optimize developmental outcome. No particular anti-epileptic medication was found to be consistently more effective in the eight families with seizure disorder, although three individuals in families F and O had significant improvement with the addition of lamotrigine that inhibits voltage-sensitive sodium channels and is thought to inhibit the pre-synaptic release of glutamate, which may have an additional neuroprotective effect.37 Although no currently used medication is known to specifically activate the CIC-4 transporter,38 better understanding of the underlying pathophysiology of CLCN4-related disorder may allow targeted treatment.
The growing number of clinical cases with variants that very likely affect CLCN4’s function is striking, given how recently this condition was first described6 and confirmed as a clinical condition.5 We recommend that CLCN4 be included in panels for the investigation of undiagnosed epilepsy and ID for both males and females.
References
Verkman AS, Galietta LJ . Chloride channels as drug targets. Nat Rev Drug Discov 2009; 8: 153–171.
Jentsch TJ . Discovery of CLC transport proteins: cloning, structure, function and pathophysiology. J Physiol 2015; 593: 4091–4109.
Scheel O, Zdebik AA, Lourdel S, Jentsch TJ . Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 2005; 436: 424–427.
Picollo A, Pusch M . Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 2005; 436: 420–423.
Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer AP et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry 2016; 21: 133–148.
Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013; 54: 1270–1281.
Yu L, Sawle AD, Wynn J, Aspelund G, Stolar CJ, Arkovitz MS et al. Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum Mol Genet 2015; 24: 4764–4773.
van Slegtenhorst MA, Bassi MT, Borsani G, Wapenaar MC, Ferrero GB, de Conciliis L et al. A gene from the Xp22.3 region shares homology with voltage-gated chloride channels. Hum Mol Genet 1994; 3: 547–552.
Okkenhaug H, Weylandt KH, Carmena D, Wells DJ, Higgins CF, Sardini A . The human ClC-4 protein, a member of the CLC chloride channel/transporter family, is localized to the endoplasmic reticulum by its N-terminus. FASEB J 2006; 20: 2390–2392.
Claes S, Vogels A, Holvoet M, Devriendt K, Raeymaekers P, Cassiman JJ et al. Regional localization of two genes for nonspecific X-linked mental retardation to Xp22.3-p22.2 (MRX49) and Xp11.3-p11.21 (MRX50). Am J Med Genet 1997; 73: 474–479.
Raynaud M, Gendrot C, Dessay B, Moncla A, Ayrault AD, Moizard MP et al. X-linked mental retardation with neonatal hypotonia in a French family (MRX15): gene assignment to Xp11.22-Xp21.1. Am J Med Genet 1996; 64: 97–106.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013; 369: 1502–1511.
Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 2015; 97: 457–464.
Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015; 385: 1305–1314.
Hu H, Wrogemann K, Kalscheuer V, Tzschach A, Richard H, Haas SA et al. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J 2009; 3: 41–49.
Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009; 41: 535–543.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J . A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–315.
Feng L, Campbell EB, Hsiung Y, MacKinnon R . Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 2010; 330: 635–641.
Fong P, Rehfeldt A, Jentsch TJ . Determinants of slow gating in ClC-0, the voltage-gated chloride channel of Torpedo marmorata. Am J Physiol 1998; 274 (4 Pt 1): C966–C973.
Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T . ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J 2011; 30: 2140–2152.
Mohammad-Panah R, Harrison R, Dhani S, Ackerley C, Huan LJ, Wang Y et al. The chloride channel ClC-4 contributes to endosomal acidification and trafficking. J Biol Chem 2003; 278: 29267–29277.
Suzuki H . Protein-protein interactions in the mammalian brain. J Physiol 2006; 575 (Pt 2): 373–377.
Grantham R . Amino acid difference formula to help explain protein evolution. Science 1974; 185: 862–864.
Brunklaus A, Ellis R, Reavey E, Semsarian C, Zuberi SM . Genotype phenotype associations across the voltage-gated sodium channel family. J Med Genet 2014; 51: 650–658.
Zweier C, Rittinger O, Bader I, Berland S, Cole T, Degenhardt F et al. Females with de novo aberrations in PHF6: clinical overlap of Borjeson-Forssman-Lehmann with Coffin-Siris syndrome. Am J Med Genet C Semin Med Genet 2014; 166C: 290–301.
Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MR et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am J Hum Genet 2015; 97: 343–352.
Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y et al. Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron 2014; 81: 103–119.
Lau A, Tymianski M . Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 2010; 460: 525–542.
Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hübner CA et al. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci 2007; 27: 6581–6589.
Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013; 12: 659–668.
Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 2001; 29: 185–196.
Poët M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci USA 2006; 103: 13854–13859.
Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J 2005; 24: 1079–1091.
Dierssen M, Ramakers GJ . Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav 2006; 5 (Suppl 2): 48–60.
Rickheit G, Wartosch L, Schaffer S, Stobrawa SM, Novarino G, Weinert S et al. Role of ClC-5 in renal endocytosis is unique among ClC exchangers and does not require PY-motif-dependent ubiquitylation. J Biol Chem 2010; 285: 17595–17603.
Nguyen DK, Yang F, Kaul R, Alkan C, Antonellis A, Friery KF et al. Clcn4-2 genomic structure differs between the X locus in Mus spretus and the autosomal locus in Mus musculus: AT motif enrichment on the X. Genome Res 2011; 21: 402–409.
Tufan K, Oztanir N, Ofluoglu E, Ozogul C, Uzum N, Dursun A et al. Ultrastructure protection and attenuation of lipid peroxidation after blockade of presynaptic release of glutamate by lamotrigine in experimental spinal cord injury. Neurosurg Focus 2008; 25: E6.
Adkins GB, Curtis MJ . Potential role of cardiac chloride channels and transporters as novel therapeutic targets. Pharmacol Ther 2015; 145: 67–75.
Acknowledgements
We thank the individuals and their families who participated in this study, and the bioinformaticians and molecular geneticists analyzing NGS data. We thank Patrick Seidler for help with oocyte measurements. We thank Professor David Sillence for his valuable assistance in the preparation of the manuscript. Part of this study was financed by a grant of the German Ministry of Education and Research through the MRNET and the EU FP7 project GENCODYS, grant number 241995; TJJ was supported by the Deutsche Forschungsgemeinschaft (SFB740); JG received NHMRC Grants 628952 and 1041920, and WKC received grant support from the Simons Foundation and National Institute of Health grant (HD057036) and was supported in part by Columbia University’s Clinical and Translational Science Award (CTSA), grant (UL1 RR024156). The DDD Study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the authors and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
WK Chung is a consultant for BioReference Laboratories. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Palmer, E., Stuhlmann, T., Weinert, S. et al. De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol Psychiatry 23, 222–230 (2018). https://doi.org/10.1038/mp.2016.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.135
This article is cited by
-
Genotype-phenotype correlation in CLCN4-related developmental and epileptic encephalopathy
Human Genetics (2024)
-
Sub-region analysis of DMD gene in cases with idiopathic generalized epilepsy
neurogenetics (2023)
-
Functional and clinical studies reveal pathophysiological complexity of CLCN4-related neurodevelopmental condition
Molecular Psychiatry (2023)
-
Systematic analysis and prediction of genes associated with monogenic disorders on human chromosome X
Nature Communications (2022)
-
Novel CLCN4 variant associated with syndromic X-linked intellectual disability in a Chinese girl: a case report
BMC Pediatrics (2021)
