
Abstract
X-linked intellectual disability (XLID) is a clinically and genetically heterogeneous disorder. During the past two decades in excess of 100 X-chromosome ID genes have been identified. Yet, a large number of families mapping to the X-chromosome remained unresolved suggesting that more XLID genes or loci are yet to be identified. Here, we have investigated 405 unresolved families with XLID. We employed massively parallel sequencing of all X-chromosome exons in the index males. The majority of these males were previously tested negative for copy number variations and for mutations in a subset of known XLID genes by Sanger sequencing. In total, 745 X-chromosomal genes were screened. After stringent filtering, a total of 1297 non-recurrent exonic variants remained for prioritization. Co-segregation analysis of potential clinically relevant changes revealed that 80 families (20%) carried pathogenic variants in established XLID genes. In 19 families, we detected likely causative protein truncating and missense variants in 7 novel and validated XLID genes (CLCN4, CNKSR2, FRMPD4, KLHL15, LAS1L, RLIM and USP27X) and potentially deleterious variants in 2 novel candidate XLID genes (CDK16 and TAF1). We show that the CLCN4 and CNKSR2 variants impair protein functions as indicated by electrophysiological studies and altered differentiation of cultured primary neurons from Clcn4−/− mice or after mRNA knock-down. The newly identified and candidate XLID proteins belong to pathways and networks with established roles in cognitive function and intellectual disability in particular. We suggest that systematic sequencing of all X-chromosomal genes in a cohort of patients with genetic evidence for X-chromosome locus involvement may resolve up to 58% of Fragile X-negative cases.
Similar content being viewed by others


Introduction
Intellectual disability (ID), which affects 1–2% of the general population, is characterized by significant sub-average cognitive functioning, commonly defined by an IQ of lower than 70, and deficits in adaptive behavior, such as social and daily-living skills with an onset before 18 years of age. Most severe forms have a single genetic cause, and males are more often affected than females. Therefore, for many years, research has focused on the molecular elucidation of X-linked forms of ID which are thought to account for 10–12% of all males with ID.1 Until 2007, mutations in XLID genes known at that time had been detected in 42% of the Fragile X-negative families studied.2 Afterwards, a large-scale, comprehensive Sanger sequencing study was performed to identify the missing genes and mutations in a cohort of 208 families.3 This study was complemented by high-resolution array CGH profiling on the same set of families4 and by further genetic and functional evidence for some of the unique missense variants.5, 6, 7 However, in excess of 50% XLID families remained without plausible gene defects further indicating genetic heterogeneity of XLID. Since then, several novel XLID genes have been reported in the medical literature, including HUWE1 [MIM 300697],8SLC9A6 [MIM 300231],9PCDH19 [MIM 300460],10RAB39B [MIM 300774],11HDAC8 [MIM 300269],12HCFC1 [MIM 300019],13CCDC22 [MIM 300859],14, 15USP9X [MIM 300072],6PIGA [MIM 311770],16WDR45 [MIM 300526],17KDM6A [MIM 300128],18BCAP31 [MIM 300398],19ZC4H2 [MIM 300897],20KIAA2022 [MIM 300524]21 and MID2 [MIM 300204].22
In this study, we aimed to (i) identify the molecular causes of XLID in a large group of unresolved families, (ii) define the number of XLID genes that can be identified by performing targeted sequencing of all X chromosome-specific exons, (iii) gain knowledge about ID-related pathways and networks and (iv) estimate the proportion of families with XLID that can be solved using X-exome sequencing. For this, we initially focused on 248 families collected by the EUROMRX consortium and associated groups that remained unresolved by pre-screening for mutations in selected known XLID genes and by array CGH. In follow-up work we investigated an additional cohort of 157 similarly pre-screened families. We took advantage of next-generation sequencing (NGS) technology to substantially improve the coverage of X-chromosomal coding sequences compared with previous studies. We identified likely pathogenic variants in a range of previously established XLID genes as well as several novel and candidate XLID genes.
Subjects and methods
Subjects
All index cases had a normal karyotype, were negative for FMR1 repeat expansion, and in most of these large indels had been excluded using array CGH. The study was approved by all institutional review boards of the participating institutions, and written informed consent was obtained from all participants or their legal guardians.
Methods
For each family, DNA from one affected male was used for constructing a sequencing library using the Illumina Genomic DNA Single End Sample Prep kit (Illumina, San Diego, CA, USA). Enrichment of the X-chromosomal exome was then performed for each library using the Agilent SureSelect Human X Chromosome Kit (Agilent, Santa Clara, CA, USA), which contains 47 657 RNA baits for 7591 exons of 745 genes of the human X chromosome. Single-end deep sequencing was performed on the Illumina Genome Analyzer GAIIx (Illumina, San Diego, CA, USA). Read length was 76 nucleotides. For a subset of families of the second cohort, we performed droplet-based multiplex PCR (7367 amplicons, 757 genes, 1.54 Mb) similarly to the previously described study.23 Paired-end deep sequencing was performed on the HiSeq2000 platform (ATLAS, Berlin, Germay). A scheme outlining the variant discovery workflow is presented in Supplementary Figure 1.
Reads were extracted from qseq-files provided by the Illumina GAII system (Illumina). Reads containing ambiguous base calls were not considered for further analysis. The remaining reads were subsequently mapped to the human reference genome (hg18 without random fragments) with RazerS24 (parameters: -mcl 25 -pa -m 1 -dr 0 -i 93 -s 110101111001100010111 -t 4 -lm) tolerating up to 5 bp differences to the reference sequence per read. Only unique best matches were kept, whereas all remaining reads and those containing indels were subjected to a split mapping procedure of single end reads (SplazerS version 1.0,25 parameters: -m 1 -pa -i 95 -sm 23 -s 111001110011100111 -t 2 -maxG 50000) to detect short insertions (⩽30 bp) and larger deletions (<50 kb). For detecting large insertions/deletions by analyzing changes in depth of coverage along the targeted regions we used ExomeCopy.26 We performed a quality-based clipping of reads after mapping but before calling variants to minimize the number of false-positive calls. Starting from each end of a read with a sliding window of 10 bp we trimmed the read until we observed a window with all 10 phred base quality values >10. If there was a variant within 3 bp distance to the clipped region then the trimming was expanded up to this potential sequencing error. For both mapping procedures (RazerS+SplazerS) the calling of a variant required at least three reads with different mapping coordinates to exclude potential amplification artifacts. Single-nucleotide polymorphisms (SNPs) and short indels (⩽5 bp) were called with snpStore (parameters: -reb 0 -fc 10 -m 1 -mmp -mc 3 -oa -mp 1 -th 0.85 -mmq 10 -hr 0.001 -re -pws 1000), performing a realignment of the clipped mapped reads whenever at least three indel-containing reads were observed within close proximity. For an indel to be called no more than 75% of the spanning reads were allowed to contradict it. For single base variants we used the Maq consensus statistics27 integrated into the snpStore code. Larger deletions and small insertions were identified by examining the split mapping results for potential breakpoint positions. In case of multiple such positions implying varying indel lengths within a 20-bp range such candidate calls were assumed to be unreliable and were therefore discarded. To detect potential retrocopies, the boundaries of split read mappings were compared with known exon boundaries allowing a tolerance of ±5 bp. When both split ends coincided with exon boundaries these exons were defined as being part of a retrocopy event. Completeness of the retrocopy was defined by the highest fraction of exons per transcript for which exon-spanning reads were detected. One example is shown in Supplementary Figure 2. In a parallel approach, we processed the sequencing reads using an alternative software, Medical Resequencing Analysis Pipeline (MERAP), for mapping, variant calling, and annotation.28 Here, the mapping was performed using SOAP2.2029 allowing at most two mismatches. For the calling of single-nucleotide variants (SNVs) and indels a minimum of four reads and a more stringent Phred-like quality score of ⩾20 were required. Finally, only those variants called by both approaches were kept to yield high-confidence candidate variants.
For in silico prioritization of variants, we integrated the following features: (a) gene/transcript annotations (downloaded from UCSC Genome Browser, hg19); (b) known sequence variants from the following data sources: dbSNP, 1000 Genomes project, 200 Danish exomes,30 NHLBI Exome Sequencing Project (ESP6500, version without indels). Base exchanges were considered as 'known' (with exception of SNVs observed as only heterozygous in ESP6500 and 1000 Genomes project) if position and type of the nucleotide were identical to entries in the reference databases. We did not use a cutoff based on minor allele frequency. In case of short indels, a tolerance in positional matching was applied based on repetitiveness of the deleted/inserted sequence in the SNV flanking sequence; (c) variants detected in the screen performed by Tarpey et al.3 were located in transcripts derived from ENSEMBL version 54. We defined the amino-acid coordinate shared by most transcripts of a gene as reference, which is sometimes different from the one annotated by Tarpey et al.3 Conversion of coordinates was successful for 1647 variants; (d) evolutionary conservation across 44 vertebrate species;31 (e) splice site detection for defining potential cryptic splice sites (software NNSplice; cutoff 0.9 (ref. 32)); (f) potential functional impact: PolyPhen2,33 SIFT34 and (g) Human Gene Mutation Database (HGMD): known variants with Pubmed entries were treated as potentially disease causing if they were listed in HGMD Professional and annotated in maximally one reference SNV database.
We thus defined a prioritization score (PS) based on basic, computationally tractable criteria like type of variant or evolutionary conservation. Polyphen2/SIFT produces a categorical output (benign/tolerated, possibly damaging/low confidence, probably damaging/damaging), which was assigned to ordinal variables 1, 2 or 3. Numbers are decreasing with decreasing functional impact, missing values are scored nil. Whenever only one of the methods scored >0, the zero score was set to 1 to avoid underestimation of the functional impact. PhyloP values were rounded down to decimal numbers, values >5 were set to PHY=5, for values <2 PHY= 1, for values <0 PHY= 0. Since deletions/insertions are usually not scored by PolyPhen2/SIFT, we defined the following adhoc weighting scheme: non-sense/frameshift: TYPE=20 (maximal PS), deletions (>50 bp): TYPE=9 (similar to maximal impact prediction by PolyPhen2 and SIFT), duplications, in-frame deletions, potential splice site variants: TYPE=3. The score for a change identified in a gene known to have a role in XLID before this study was set to 3. PS=PP2 * Sift+PHY+TYPE+XLID; if PS>20, PS=20.
We also used CADD (Combined Annotation-Dependent Depletion)35 as an additional tool for annotating and interpreting SNVs as well as small indels (see Supplementary Figure 3 for comparison of the scores).
Analysis of human CLCN4 SNVs in Xenopus oocytes
CLCN4 SNVs were introduced into human CLCN4 (NM_001830.3; Gene ID: 118) cDNA cloned into pTLN and pCIneo36 by recombinant PCR. We assessed the expression level and stability of wild-type and mutants with p.Gly78Ser, p.Leu221Val, p.Val536Met or p.Gly731Arg substitutions by western blot analysis of lysates from transiently transfected cells using standard methods. Xenopus laevis oocytes were injected with 23 ng cRNA, which was transcribed with the mMessage Machine kit (Ambion, Thermo Fisher Scientific Inc., Waltham, MA, USA) from pTLN.37 After 3 days incubation at 17 °C, currents were measured at room temperature using standard two-electrode voltage clamp employing TurboTEC amplifiers (npi electronic, Tamm, Germany) and pClamp10.2 software (Molecular Devices, Sunnyvale, CA, USA). Oocytes were superfused with modified ND96 saline (96 mM NaCl, 2 mM K-gluconate, 1.8 mM Ca-gluconate, 1 mM Mg-gluconate, 5 mM HEPES pH 7.5) and clamped in 20-mV steps to voltages between −100 and +80 mV. The holding potential was −30 mV.
Morphological studies of mouse hippocampal neurons
Mouse embryos were dissected at embryonic day 16.5 (E16.5), tissue was dissociated by trypsin as well as by mechanical treatment, and primary cultures of hippocampal neurons were established at 37 °C by plating on coated-glass coverslips (poly-L-Lysine and Laminin) at a density of 100 000 per 16 mm Petri dish. Neurons were differentiated for 18 days in vitro (18 DIV) using Neurobasal/B27 medium and antibiotics (Mycozap, Lonza, Basel, Switzerland), replacing half of the media each third day for maintenance according to standard procedures.38, 39 Short-hairpin RNA (shRNA) design was made by targeting the 3′UTR of each specific gene using Promega shRNA designer tools (Promega BioSciences, San Luis Obispo, CA, USA) or informations based on The RNAi Consortium (TCR) shRNA Library. A control shRNA-producing plasmid was used in control experiments as previously described.40 Three independent shRNA-producing and GFP-expressing plasmids, based on pSystrike vector (Promega), were produced for each gene and used as a pool. Sequences targeted by shRNAs for Cnksr2 and Clcn4–2 (Clcn4) genes and control sequences are GGAGCAGAGGATGGCAGTCATTCA, GGTGGGAAGGCTAGCTCTGTTACT, GCGCGGCGTATCAGGGCAAAGCTT, GGGTATGTGGGAGGGTGTAAATGA, GGGAGAGGCGAGTACGAAGATGAA, GTGGTCTACTCATGGCCATCTCAT and GCTCACCCTTCCTACTCTC. Full-length murine Cnksr2 (NCBI reference sequence NM_177751.2) and Clcn4 (NCBI reference sequence NM_011334.4) cDNAs were cloned into pFN21A HaloTag® CMV Flexi® vector (Promega). For rescue experiments, pool of plasmids encoding shRNAs were cotransfected with pFN21A HaloTag® fused to either Cnksr2 or Clcn4. All constructs were sequence verified and plasmids were purified using an endotoxin-free kit (Macherey Nagel, Düren, Germany). Transfections were carried out using Lipofectamine (Invitrogen, Life Technologies, Carlsbad, CA, USA) at 11 DIV and cells were fixed at a later stage of differentiation for analysis (DIV 18). Individual neurons were directly imaged under fluorescence and confocal microscopy (spinning disk microscope, Leica, Leica Microsystems, Wetzlar, Germany) using GFP labeling as a tracer of morphology. Immunocytochemistry to detect GFP (goat antibody, Abcam, Cambridge, UK) and HaloTag constructs (HaloTag TMR Ligand, Promega) was also realized according to standard procedures. Image analysis was done using Imaris software with ‘Filament tracer’ plugin (Bitplane Scientific Software, Bitplane AG, Zürich, Switzerland) and ImageJ software (Wayne Rasband, Bethesda, NIH). It allowed quantifying total arborization of neurites (dendrites and axon) for each neuron (total length of neurites, numbers of branches, branching complexity), see Supplementary Figure 4 for details on branching analysis. Quantification was based on three independent experiments with more than 15 cells of each type per experiment analyzed. Mann-Whitney statistical test was used to compare total neuritic length as well as number of branches, whereas Chi-square test was used to evaluate significance of variations in branching complexity. Primary cultures of hippocampal neurons from wild-type and Clcn4−/− mice41 were obtained as described above with some minor differences. Animals were dissected at postnatal day 1 (P1), papain was used for dissociation of tissue, and glass coverslips were coated with poly-L-Lysine and collagen. At DIV 11, neurons were transfected with pEGFP-C1 vector (Clontech, Mountain View, CA, USA) using Lipofectamine-2000 according to the manufacturer’s instructions. Cells were fixed and stained at DIV15 or DIV18 as described previously.42 Primary antibodies were chicken anti-GFP (Aves Lab, Tigard, Oregon, USA) and mouse anti-microtubule associated protein 2 (Chemicon/Millipore, Merck/Millipore, Darmstadt, Germany) as a neuronal marker. Secondary antibodies conjugated to Alexa Fluor 488 or 546 were from Molecular Probes. Images were taken using a LSM510 laser scanning confocal microscope equipped with a × 10 lens (Zeiss). Image analysis was performed in a blinded manner using ImageJ and its plugin NeuronJ: ns (non-statistically different), *P<0.05, **P<0.01, ***P<0.001 for validation.
Results
Initially, using genome partitioning and NGS we investigated a cohort of 248 unresolved families with suggestive X-chromosome involvement. Each of the families has at least 2 affected males and in 210 families affected males were present in separate sibships. Before this study, 125 of the index patients had been prescreened for different (per case) known XLID genes.2, 23 For 1/3 of the 248 families, linkage data were available. For enrichment, we used probes covering 745 X-chromosome genes, including 1 224 575 bp in coding regions and 2 400 136 bp in exonic regions. In all, 92% of the target sequences were covered by at least three sequence reads, and 94.2% were covered by at least one read (Supplementary Figures 5, 6, 7 and 8). In total, we identified 3378 recurrent and 1299 non-recurrent exonic variants, which were also found in control populations (Table 1). After filtering against variants from >7000 controls, present in publicly available databases as well as our in-house database, 28 recurrent and 765 non-recurrent exonic variants, as well as 16 potential splice site variants remained (Tables 1, 2, 3, Supplementary Tables 1, 2, 3 and 4).
As a follow-up study we investigated an additional cohort of 157 unresolved, similarly pre-screened XLID families. For this cohort, we present data on pathogenic variants identified in known XLID genes, likely or potentially pathogenic variants in novel and candidate XLID genes and truncating variants unlikely implicated in XLID.
For validation and segregation analyses, we prioritized variants by defining a prioritization score (PS). This score incorporates several computationally tractable pieces of information including the type of variant, evolutionary conservation and (if available) evidence that the gene has a role in XLID. Except for duplications and small in-frame indels, variants with a PS of ⩾5 were considered as strong candidates for follow-up studies. More recently, we also assigned C-scores obtained by applying CADD35 for ranking SNVs and short indels.
Pathogenic variants identified in known XLID genes
A critical survey of the medical literature suggests that there are currently ~90 well-established XLID genes (79 previously known genes proposed as ‘confirmed’ by Piton et al.43 plus HCFC1,13MAOA,44CCDC22,14USP9X,6PIGA,16WDR45,17KDM6A,18BCAP31,19ZC4H2,20KIAA2022,21MID222). In these genes, we identified likely pathogenic variants in 39 of the 248 families (16%) and in 16 of the 157 families (10%), which together with the 24 families from these two cohorts that were resolved through this screen and were published earlier,7, 20, 21, 23, 45, 46, 47, 48, 49, 50 account for 21 and 18% of the cohorts (for details, see Supplementary Tables 5 and 6). The variants include co-segregating protein truncating variants, in-frame deletions or missense changes and none of them were reported in 61 486 unrelated individuals (ExAC Browser). According to the current literature and HGMD (as of May 2014), for some of these XLID genes only very few families with pathogenic variants have so far been reported, suggesting that mutations in these genes are very rare. One example is ACSL4 (previously known as FACL4 [MIM 300157]), which has a role in long-fatty-acid metabolism. Its involvement in XLID was discovered more than 10 years ago.51 Yet, until today a total of only four unrelated families with pathogenic point variants in ACSL4 have been published. These include one recurrent amino-acid change and one splicing variant.51, 52, 53 We identified a non-sense variant in this gene (chrX:108902601G>A, p.Arg654*) present in one family (Supplementary Table 5). Our results strongly support that ACSL4 mutations cause XLID. In NLGN3 [MIM 300336], that is known as ‘autism’ gene with currently two likely pathogenic missense and two potentially disease relevant splicing variants reported in the literature,54, 55, 56 we identified a likely deleterious stop codon (p.Arg162*) which is expected to remove most part of the protein. The variant could not be tested for segregation because additional family members were unavailable. The 29-year-old affected is the first and only child of unrelated and healthy parents. He presented with moderate ID, severe behavioral problems, especially abnormal sexual behavior and aggression. There was no formal diagnosis of autism. We have also identified pathogenic variants in ID genes with widely varying phenotypes, which are difficult to diagnose by clinical examination alone. One example is PQBP1 [MIM 300463] in which we found a pathogenic single nucleotide deletion that causes a frameshift resulting in a premature stop codon (p.Phe240Serfs*26, Supplementary Table 5). As a result of this molecular diagnosis, careful reexamination of the affected boys revealed subtle dysmorphic features that are in good agreement with the currently known PQBP1 clinical spectrum.
One of the established XLID genes with several likely pathogenic variants identified in our study groups is MED12 [MIM 300188]. Missense variants in this gene have been linked with Lujan-Fryns syndrome57 [MIM 309520], Opitz-Kaveggia syndrome58 [MIM 305450] and Ohdo syndrome59 [MIM 300895]. In addition to the previously published large family with a protein truncating variant associated with a profound phenotype in males and several heterozygous female carriers with variable cognitive impairment,28 we identified segregating likely pathogenic missense variants in three families (Supplementary Table 5). Similarly, eight XLID families carry pathogenic variants in CUL4B [MIM 300304] (Supplementary Table 5).50
Likely pathogenic variants in novel XLID genes and previously proposed candidate genes
We identified likely deleterious variants in four novel XLID genes and validated three previously suggested candidates, described here in more detail, including CLCN4 [MIM 302910], CNKSR2 [MIM 300724], FRMPD4 [MIM 300838], KLHL15, LAS1L, RLIM [MIM 300379] and USP27X. We propose these genes to be confirmed or novel X-chromosome ID genes based on our genetic, bioinformatic and functional evidence as well as current knowledge extracted from the literature. All but one variant identified in these genes co-segregated with ID in the relevant families (Table 2, Figures 1 and 2).

Apparently pathogenic CLCN4 mutations identified in the screen and functional analysis of the missense variants. (a) Pedigrees of families with CLCN4 likely pathogenic mutations. Individuals tested for co-segregation with X-linked intellectual disability (XLID) and the results are indicated, *=mutation carrier, wt=subject does not carry the mutation. (b) Current–voltage relationships of the electrogenic Cl−/H+ exchanger protein ClC-4 and its mutants expressed in Xenopus oocytes, shown as mean values of normalized steady-state currents from several oocytes (numbers indicated in figure, in parentheses: number of frogs). Compared with the strongly outwardly-rectifying currents of wild-type ClC-4,36, 121 currents were much smaller or even absent with CIC-4 mutant proteins carrying p.Gly78Ser, p.Leu221Val, p.Val536Met and p.Gly731Arg substitutions. ctr, non-injected controls; error bars, s.e.m. Two-tailed t-test was used for statistical comparisons (**P<0.01, ***P<0.001 compared with wild-type ClC-4 currents). (c) Analogous positions of amino acids mutated in ClC-4 highlighted in the crystal structure of CmClC.63 Amino acids are displayed as spheres in colors like in (b). The small green spheres represent Cl− ions. CLC transporters form dimers of identical subunits (shown in different shades) and include a transmembrane domain (TMD) and two cytosolic cystathionine-β-synthase (CBS) domains.

Pedigrees of families with co-segregating truncating and missense variants in novel and previously suggested candidate X-linked intellectual disability (XLID) genes validated through this study. (a) In the postsynaptic density protein CNKSR2, we observed a protein truncating variant in family P180. (b) In FRMPD4, we detected a unique protein truncating variant in family P58 with five affected males. (c) In KLHL15, we identified a protein truncating variant in family D60 with eight affected males. (d) In LAS1L, we found unique missense variants in families MRXS6 (ref. 66) and T50, both with Wilson-Turner (WTS) syndrome. (e) In RLIM, we identified missense variants in three large families D72, T11 and AU31. (f) In USPX27, we found a protein truncating variant in family D177 and a missense variant in family L75. (g) In the novel candidate XLID gene CDK16, we detected a protein truncating variant in family L56. (h) In the novel candidate XLID gene TAF1, we identified missense variants in families D185 and N67. *=mutation carrier, wt=wild type.
In CLCN4, that encodes the electrogenic chloride/proton exchanger ClC-4,60 we discovered a protein truncating variant (p.Asp15Serfs*18, family MRX4961) and four missense variants (p.Gly731Arg, family MRX15,62 p.Leu221Val, p.Val536Met, p.Gly78Ser) (Figure 1a). ID of the affected males was variable, even within families, ranging from mild to severe. Similarly, intra- and interfamilial variable clinical features include epilepsy, dysmorphic face, scoliosis and strabismus (for detailed clinical information, see Supplementary Table 7). All affected amino-acid residues lie within the transmembrane part or in the cytoplasmic, carboxy terminus of the protein (Figure 1c). To provide further evidence that the missense variants identified impair ClC-4 protein function, we performed analyses in Xenopus laevis oocytes. Compared with the strong outwardly-rectifying currents of wild-type CLC-4 (refs. 36, 60) currents were much smaller or even absent with ClC-4 constructs carrying the point variants, showing that these substitutions markedly impaired the function of the ClC-4 protein (Figure 1b). In the crystal structure of algal CmClC,63 p.Gly731 is located just at the contact sites of the cytosolic cystathionine-β-synthase (CBS) domains of the different subunits of the ClC-4 homodimer. Since CBS domains have been implicated in the gating of CLC channels,64, 65 the p.Gly731Arg substitution may interfere with this process. We additionally analyzed the effects of the mouse counterpart, Clcn4, on neuronal differentiation by transfecting hippocampal neurons at day-in-vitro 11 (DIV11) with knock-down constructs targeting this gene and evaluated the cells at a later stage of differentiation (DIV18). At this stage, neuronal differentiation is complete and was clearly affected in Clcn4-depleted cells. Indeed, compared with controls that were transfected with a non-silencing construct, in Clcn4 depleted cultures neurons were less branched, that is, the total length of neuritic branches was decreased by 30% corresponding to less dendritic branches per cell. However, there was no effect on the complexity of dendritic branching. Introduction of ClC-4 protein in knock-down cells using RNAi-insensitive cDNA rescued both dendritic phenotypes to control levels, thus highlighting the specificity of the phenotype truly associated with the loss of ClC-4 protein (Figure 3a). Primary neurons derived from Clcn4−/− mice41 confirmed these findings (Figure 3b). Although the observed morphological changes were more subtle when compared with those obtained with the shRNA-mediated knock-down, they were statistically significant.

Effects of Clcn4 or Cnksr2 downregulation on morphology of mouse hippocampal neurons. Typical arborization of GFP-labeled neurons cultured for 18 days in vitro (DIV) after targeting by non-silencing (NS) or gene-specific shRNA (Clcn4 or Cnksr2) at 11 DIV. Quantification of transfected neurons, for total length of neuritic branches, total number of branches (a branch is considered as the segment between two branching points) and for dendritic branching complexity (levels were quantified per neuron from 1 to 6, each time a branching point is met from nucleus toward the distal part of each dendrite). Detection of co-transfections of shRNA and cDNA encoding plasmids for rescue experiments is shown as overlap of GFP (green) and Halotag (red) signals. Clcn4 experiment is shown in (a) and Cnksr2 in (c). More than 15 representative cells of each type were analyzed per experiment, with three independent experiments conducted. (b) Quantification of neuritic arborization in GFP expressing primary hippocampal neurons derived from Clcn4+/+ and Clcn4−/− mice as described above. Two independent experiments with>30 cells per genotype of five wild-type and four knock-out mice were analyzed. ClC-4-deficient neurons showed a significant reduction in the total number and total length of neuritic branches compared with wild-type cells. Average values with s.e.m. are shown (i) in histograms for neuritic length and number of branches and (ii) in curves for complexity levels of branching. Mann-Whitney and Chi2 tests were respectively used for statistical comparisons (ns: non-statistically significant, *P<0.05, **P<0.01, ***P<0.001). Scale bar represents 10 μm.
In CNKSR2 (also known as CNK2, KSR2, MAGUIN), we identified a likely pathogenic frameshift variant (p.Asp152Argfs*8) in a family with four affected males. This variant was present in three affected brothers tested and their mother (Figure 2a). That CNKSR2 is implicated in ID is further supported by an unrelated intellectually impaired female who carries a balanced translocation with a chromosomal breakpoint that disrupts CNKSR2 (J Chelly et al., unpublished result). To assess whether the loss of the mouse ortholog, Cnksr2, has a functional impact, we depleted it in primary hippocampal neurons fully differentiated in vitro. Reduction of Cnksr2 had a profound effect on the number of dendritic branches, as well as on total length of neurites per neuron, which were all reduced by 65–75% (Figure 3c). These two drastic phenotypes were partially, but highly significantly rescued in neurons by expression of a shRNA-resistant cDNA plasmid encoding HaloTag-fused Cnksr2 protein. Furthermore, dendritic branching complexity was largely affected due to loss of terminal branches (level 4 30%, level 5 50% and level 6 70%). This phenotype could be restored in the rescue experiment (Figure 3c).
In FRMPD4 we identified a protein truncating variant (p.Cys618Valfs*8) in a single XLID family with five affected males in different sibships (Figure 2b) and a de novo missense mutation in an unrelated male who at the age of 17 years presented with significant developmental delay, the absence of speech and autism spectrum disorder. His brother and a half-brother did not carry the mutation, and upon re-examination it appeared that they had much milder phenotypes characterized by learning problems at school.
In the poorly characterized KLHL15 gene, encoding a member of the kelch-like protein family, we identified a protein-truncating variant (p.Tyr394Ilefs*61) that co-segregates with ID in a large family with eight affected males in three different sibships (Figure 2c). Further support for KLHL15 being implicated in XLID comes from an unrelated XLID family, which carries a small deletion that removes part of KLHL15 and is expected to result either in a C-terminally truncated protein or in a complete loss of KLHL15 (J Gecz, V Kalscheuer and F McKenzie et al., unpublished result).
Two likely causative missense variants potentially affecting protein biosynthesis or transcription regulation involved LAS1L, encoding the human homolog of the highly evolutionary conserved S. cerevisiae protein Las1 (lethal in the absence of SSD1-v1). The p.Ala269Gly substitution was identified in the large original family described as Wilson-Turner syndrome (WTS, MIM 309585) with mild to moderate ID and obesity (Figure 2d). 66 More than half of the affecteds had speech disability (mutism or stuttering), small or undescended testes and relatively small feet. The p.Arg415Trp substitution was present in an unrelated family from France with five affected males in three different sibships (Figure 2d). Upon clinical re-examination of affected males from this family, they all turned out to have ID with speech impairment, obesity and hypogonadism, too.
For RLIM, which encodes the RING-H2 zinc finger protein 12, we identified three families who carry unique missense variants that resulted in single amino-acid substitutions (D72, p.Arg387Cys; T11/MRX61, p.Pro587Arg; AU31, p.Arg599Cys). All variants co-segregated with XLID in these large families (Figure 2e) and affected highly conserved amino-acid residues. Both p.Pro587Arg and p.Arg599Cys substitutions affect amino acids of the zinc-finger domain of RLIM. In addition, HOPE67 predicts that the differences in amino-acid properties disturb this domain.
Family D177 with three affected males in different sibships carries a 5-bp deletion (g.AAGTA) in USP27X encoding ubiquitin-specific peptidase 27. The deletion was also present in their mothers (Figure 2f). The variant results in a frameshift and premature stop codon (p.Ser342Argfs*14) that is expected to remove the C-terminal part of the corresponding protein. The unrelated family L75 with four affected males (Figure 2f) carries a potentially deleterious missense variant in USP27X, which substitutes a highly conserved tryptophane by a histidine residue (p.Trp381His).
Variants identified in novel candidate XLID genes
In CDK16 (also frequently named in the literature as PCTK1 and PCTAIRE1, [MIM 311550]), which is highly expressed in brain and testis, we identified a dinucleotide deletion in the index male of family L56. The deletion affects all three known RNA isoforms and results in a frameshift and a premature stop codon before the N-terminal kinase domain (p.Trp326Valfs*5). The deletion was also present in his affected brother and two affected male cousins (Figure 2g) who, in addition to ID, all suffered from spastic diplegia. It is currently unclear whether another dinucleotide deletion (g.TG, chrX:47085594-47085595, p.Phe322Trpfs*12), which would truncate the C-terminus of only one CDK16 protein isoform (non RefSeq variant), and was identified in a single family is a rare neutral variant. CDK16 is a poorly characterized atypical member of the cyclin-dependent kinase family. It is particularly abundant in postmitotic neurons,68 and has been implicated in the regulation of neurite outgrowth,69 neuronal migration, vesicular transport and exocytosis.70, 71, 72 Depletion of Cdk16 abolished dendrite development in primary neuron cultures,73 and in C. elegans it is important for localizing presynaptic components.74 Thus, it is plausible to assume that loss of CDK16 function could have a role in ID, but more evidence is required to accept CDK16 as a novel XLID gene.
In TAF1 [MIM 313650] we identified segregating missense variants in two unrelated families (Figure 2h), both of which affect highly conserved amino acids of proteins encoded by the longest transcript isoforms encoding the TATA box binding protein-associated factor, 250 kD (TAF1), which is a subunit of a complex with a key role in transcription initiation. Additionally, TAF1 is part of the H3K4 methyltransferase MLL1, which also contains CHD8 that is implicated in autism.75, 76 Reduced expression of TAF1 has been shown in brain tissues from patients with X-linked Dystonia-Parkinsonism [MIM 314250], a movement disorder endemic to the Philippines.77 Furthermore, variants in TAF2 have been associated with autosomal recessive ID.78, 79 Although additional evidence for TAF1 being implicated in XLID is currently missing, these data indicate that loss of TAF1 function could affect cognition.
Variants with unlikely effect on brain function
We also identified protein truncating or read-through changes in 40 genes which we considered as unlikely to cause ID because these were (i) outside the linkage intervals in the respective families and therefore expected not to co-segregate with the phenotype, (ii) did not co-segregate with ID, (iii) previously reported in healthy males3, 80 or (iv) involved in phenotypes distinct from ID. From these, protein truncating variants that have not been reported in controls are presented in Table 3. The ARSF [MIM 300003] variant is present in a family with two affected males. Other ARSF truncations were reported in controls.3 The index patient sequenced here additionally carries a non-segregating truncating variant in MAGIX, but we currently cannot entirely rule out that this proband is a phenocopy. Single truncating variants in COL4A6 [MIM 303631], CXorf61 [MIM 300625] and MAP3K15 [MIM 300820] are outside the linkage intervals of the respective families and therefore unlikely co-segregate with XLID in the families in which they were found. Similarly, the GUCY2F [MIM 300041] and SLC25A43 [MIM 300641] truncating variants did not co-segregate with XLID in the family in which they were identified and other rare protein truncating variants were reported in ESP6500 and in other healthy male controls.81, 82 Furthermore, COL4A6 is part of a contiguous gene deletion causing Alport syndrome [MIM 301050], a childhood onset progressive haematuric glomerulopathy with high-frequency sensorineural hearing loss and typical ocular signs, and for MAP3K15 other stop-gain variants have been identified in male controls.3 CXorf64 and FATE1 [MIM 300450] truncating variants were identified in the same family and both did not co-segregate with XLID. Variants in FRMD7 [MIM 300628] cause idiopathic infantile nystagmus [MIM 310700]. Similarly, a non-segregating stop-gain variant in GPR112 and a non-segregating stop-loss variant in the XLID gene HDAC8 [MIM 300269] (Table 3) were found in a family with a co-segregating protein truncating variant in the XLID gene UPF3B [MIM 300298], which was considered as the cause of ID (Supplementary Table 5). For HS6ST2 [MIM 300545], our follow-up study revealed that this deletion is recurrent and also present in a family with a pathogenic variant in the known XLID gene KDM5C (previously known as JARID1C [MIM 314690]). Similarly, other HS6ST2 truncating variants were identified in normal males.30 Furthermore, a recurrent RAB40AL dinucleotide missense variant (p.Asp59Gly) previously reported to cause Martin-Probst syndrome83 [MIM 300519] and published as causal in an unrelated male84 was identified in four unrelated index patients and did not segregate in two of the families. In another family, a protein truncating variant was present on both X-chromosomes of healthy females, as recently reported.85
Discussion
For many years, research into the molecular causes of ID has focused on the X-chromosome, prompted by the observation that males are more often affected than females.86, 87 Cumulatively, sequencing of positional and functional candidate genes as well as high-resolution array CGH led to the identification of apparently causative defects in more than 100 X-linked genes, but after the advent of high-throughput sequencing techniques, mutations inactivating some of these genes were also observed in healthy individuals, thereby questioning the identity of several of the previously identified XLID genes.43
Despite the large number of established XLID genes, more than half of the XLID families remained unsolved,2, 3 suggesting further heterogeneity. This prompted us to investigate a cohort of 405 XLID families by NGS. 74 (18%) of the families carry variants in established XLID genes that we consider as causative. Six families (1.5%) carry potentially causative XLID variants which, in our opinion, have to be studied in more detail before qualifying for carrier testing or prenatal diagnosis. Some of these variants are recurrent and were previously reported in other XLID families (for example, ATRX, CUL4B, HUWE1, for more details see our previously unpublished variants showing the respective HGMD entries in Supplementary Table 5). We did not identify any pathogenic variants in genes with an unclear role in XLID,43 apart from a co-segregating missense variant identified in ARHGEF6 [MIM 300267] the functional relevance of which remains to be established. This does not disprove a possible role of these genes in ID.
In 5% of the families, we identified likely deleterious variants in novel XLID genes and previously proposed candidate genes. In 2% of the families, mutations were observed in XLID genes that emerged from this screen and have been or will be reported in detail elsewhere, for example, ZC4H2,20 KIAA2022,21 THOC2 [MIM 300395] (Kumar et al., manuscript in preparation) and EIF2S3 [MIM 300161] (manuscript in preparation). None of these variants was found in >61 486 ‘healthy’ controls except for 3 heterozygous females with RLIM protein truncating variants, and none of these genes carry loss-of-function variants in these controls (dbSNP138, ExAC Browser30, 81, 88, 89, 90).
One of the novel XLID genes discovered in this study is CLCN4 in which we identified protein truncating and missense variants in five unrelated families, including families MRX15 (ref. 62) and MRX49 (ref. 61) with non-syndromic XLID. Electrophysiological studies in Xenopus laevis oocytes showed that the amino-acid substitutions present in the affected males markedly impaired ClC-4 function and primary mouse neurons depleted of Clcn4, the mouse counterpart of CLCN4. As well, primary neurons derived from Clcn4 knock-out mice showed a significant effect on neuronal differentiation thereby corroborating that ClC-4 is important for cognition. Our results further support pathogenicity of a de novo CLCN4 missense variant identified in a boy with epilepsy and cognitive dysfunction.91 Very little is known about the physiological role of ClC-4. It is a member of the CLC family and most homologous to ClC-3 and ClC-5. Similar to ClC-4, other members of this family are also required for normal brain function, for example, ClC-2 variants have been described in individuals with leukoencephalopathy and MRI abnormalities,92 and loss of ClC-7 leads to neurodegeneration associated with lysosomal storage and osteopetrosis, respectively.93, 94, 95 Clcn4−/− mice do not display an obvious phenotype,41 whereas Clcn3−/− mice are developmentally retarded, show neurological manifestations and severe postnatal degeneration of the hippocampus,96 and Clcn6−/− mice display lysosomal storage in neurons.97 Thus, direct and indirect evidence point to a vital role for ClC proteins, including ClC-4, in the central nervous system.
Interestingly, the proteins encoded by the now confirmed XLID genes CNKSR2 and FRMPD4 (also termed PDZD10, PDZK10, Preso and Preso1) interact with PSD95 (Figure 4), the major scaffold protein of the postsynaptic density, which has an important role in neuronal plasticity. In CNKSR2, we identified a deleterious variant in a single family. This result was conducive to interprete a previously reported intragenic deletion identified in a boy with non-syndromic XLID98 and of two additional CNKSR2 gene deletions present in unrelated families.99 In addition, depletion of Cnksr2 in primary hippocampal neurons resulted in reduced number and complexity of dendritic branches. CNKSR2 is also connected with the XLID protein DLG3 and the ID/autism protein SHANK3 (ref. 100) is involved in the assembly of synaptic junction components,101 and modulates Rac cycling during spine morphogenesis.102

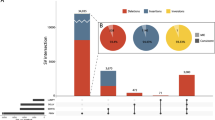
Novel X-linked intellectual disability (XLID) genes and candidates that emerged from this study encode components of key cellular protein networks. All available protein–protein interactions involving known intellectual disability (ID) proteins and the proteins likely implicated in XLID identified in this study were first extracted from the literature and then connected into a set of protein–protein interaction networks via the Ingenuity tool. Functional cellular subnetworks were extracted by using the available annotations of the interacting proteins (e.g., defined by functional category ‘translation/transcription’) and by performing literature searches. (a) PSD-95 (postsynaptic density protein 95)/Ras/Rho interaction network. CNKSR2 (CNK2, MAGUIN1, validated XLID protein) that likely functions as an adapter protein or regulator of Ras signaling pathways interacts with PSD-95 in synaptosomes.122 FRMPD4 (Preso, validated XLID protein), which is a positive regulator of dendritic spine morphogenesis and density and is required for the maintenance of excitatory synaptic transmission, interacts with PSD-95,104 and together with its binding partner ARHGEF7 (βPix) localizes in dendritic growth cones.123 (b) Transcriptional/translational interaction network. Known protein complexes are highlighted. RNA Polymerase II (RNAPII) complex with the core component TAF1 (novel candidate XLID protein). ATN1 (known ID protein) interacts with TAF4 and negatively regulates transcription of RNAPII.124 Large ribosomal subunit (60S) contains RPL10 (known candidate XLID/autism protein). LAS1L (novel XLID protein) is essential for the biogenesis of the ribosomal subunit 60S.114 Eukaryotic translation initiation factor, EIF2S3 (novel XLID protein), is a component of the translation initiation complex and promotes binding of the initiator methionyl-tRNA to the 40S ribosomal subunit.125 POLDIP3 (SKAR), involved in positive regulation of translation, associates with THOC2 (novel XLID protein) as a part of the TREX complex (functioning in mRNA export),126 with mRNA surveillance factor UPF3B (known XLID protein), as well as with a core component of the exon junction complex, EIF4A3.127 CDK16 (novel candidate XLID protein) and Synapsin 1 (Syn1, known XLID protein) were shown to interact in a membrane fraction from brain.71 Cdk16 associates with 14-3-3 zeta in Neuro-2A cells.69 Mediator complex, which functions as a transcriptional coactivator, contains MED12 (known XLID protein) and MED13L (known ID protein). NIPBL (known ID protein) is involved in loading of cohesin and associates with the mediator-cohesin complex, which interfaces gene expression and chromatin structure. Histone methyltransferase MLL2 (known ID protein) associates with a core component of Pol II, POLR2B, and activates transcription.128 Deubiquitinating enzyme USP27X (novel XLID protein) interacts with USP22 that is required for histone deubiquitination,129 and which associates together with TAF10 as part of the TBP-free TAF complex (TFTC).109 ADRA2B, G-protein coupled receptor, by interacting with EIF2B130 and 14-3-3 zeta131 links G protein-mediated signaling network and cellular control of protein synthesis. (c) Ubiquitination interaction network. KLHL15 (validated XLID protein) with a function in protein ubiquitination interacts with a component of an ubiquitin E3 ligase, CUL3.132 RLIM (novel XLID protein) is an E3 ubiquitin protein ligase113 and associates with UBE2D1.133
For FRMPD4, the first evidence for its involvement in XLID came from a duplication that likely disrupted this gene in a male with mild ID and autism.103 Depletion of the FRMPD4 ortholog in the mouse decreases spine density and excitatory synaptic transmission,104 similarly to what has been described for other proteins important for normal brain function and, when deficient result in cognitive impairment.
Four of the novel and validated XLID genes are potentially directly or indirectly implicated in the regulation of protein turnover (Figure 4). One of these is KLHL15, in which we identified a deleterious variant in a large family and a deletion that likely affects its normal function in an unrelated family (unpublished results). Our results support pathogenicity of a partial deletion of KLHL15, which has very recently been described in a single proband with severe ID, epilepsy and anomalies of cortical development.105 KLHL15 is a member of the Kelch-like proteins, many of which are adaptors for the recruitment of substrates to Cul3-based E3 ubiquitin ligases for degradation by the 26S proteasome. KLHL15-Cul3 specifically targets a brain-specific regulatory subunit of the protein phosphatase 2A (PP2A/B’ß) and thereby promotes its proteasomal degradation, resulting in the formation of alternative PP2A holoenzymes.106 PP2A/B’ß has been shown to inactivate CAMKII, which is a key mediator of long-term potentiation. Thus, aberrant turnover of PP2A/B’ß caused by KLHL15 protein-truncating variants could contribute to XLID.
Little is currently known about the functional role of the ubiquitin specific peptidase USP27X. It was among the top 50 genes with enriched expression in mouse embryonic serotonin neurons and thus may be important for serotonergic function.107 The only known interaction partner of USP27X is USP22, which has been shown to be required for glial cell and neuronal development in flies.108 It is an integral component of a Pol II coactivator complex that, in addition to its histone acetyltransferase activity, has a role in the turnover of histone modifications by specifically removing the ubiquitin moiety from histones H2A and B, and it functions as a positive cofactor for activation by nuclear receptors.109 Several previously identified ID genes code for subunits of the same complex, for example, proteins from the mediator complex, for example, MED12 and MED13L [MIM 608771], and a range of proteins that regulate transcription by modulation of the chromatin structure.110 Furthermore, variants in another member of the peptidase C19 family, USP9X, are also associated with XLID.6
Three unrelated families carry co-segregating point mutations in the E3 ubiquitin ligase RLIM, which were all predicted as disease causing.111 Two of the amino-acid substitutions lie in the C-terminal zinc finger domain and could disturb its function. RLIM has an important role in embryonic development by acting as a negative regulator of LIM homeodomain transcription factors through two distinct and complementary mechanisms: recruitment of the Sin3A/histone deacetylase corepressor complex and targeting the coactivator of LIM homeodomain proteins for degradation,112, 113 suggesting that it has critical functions in regulating associated transcriptional activity.
LAS1L in which we identified likely pathogenic missense variants in two families with a syndromic form of XLID (WTS,66 [MIM 309585]) is involved in ribosome biogenesis. It is required for the synthesis of the 60S ribosomal subunit and maturation of 28S rRNA. Depletion of LAS1L results in a p53-dependent cell-cycle arrest, defective pre-rRNA processing and failure to synthesize mature 60S ribosomal subunits.114, 115 Additionally, LAS1L is part of a large nuclear complex (Five Friend of Methylated chromatin target of protein-arginine-methyltransferase-1) that has a role in transcription regulation by affecting the sumoylation status and transactivation potential of the zinc-finger transcription factor Zbp-89,116, 117 and is a component of the CoREST1/HDAC1 corepressor complex.117 It remains to be determined which of the LAS1L functions are compromised by the missense variants. Interestingly, another missense variant in this gene has recently been identified in a boy with congenital lethal motor neuron disease,118 suggesting that LAS1L variants are associated with a variable phenotype. Similarly, a family with a phenotype resembling WTS carries a missense variant in the known XLID gene HDAC8,119 in which loss-of-function variants are associated with Cornelia de Lange syndrome (CDLS5 [MIM 300882]).12
Our investigation has led to the identification of several novel ID genes that are mutated in up to 7% of the XLID families. There are still many ID families with evidence for X-linkage that remain unresolved, including 75 families with 4 and more affected males in separate sibships connected through female carriers, suggesting several yet to be identified genes or loci on the X-chromosome involved in ID. The ‘diagnostic’ yield of 26% obtained by performing X-exome resequencing on a pre-screened cohort contrasts with our previous experience that defects in XLID genes known until 2007 account for more than half of the families screened.2 This discrepancy can be explained by the fact that in the present study, most of the families had already undergone prior FraX testing, array CGH and targeted analysis of many previously known genes. Indeed, KDM5C variants and disease-causing variants in three other most common XLID genes, namely MECP2 [MIM 300005], IL1RAPL1 [MIM 300206] and PQBP1, turned out to be strongly under-represented in the families included here (Supplementary Table 10). To estimate the diagnostic yield of sequencing all X-chromosomal exons in novel, not previously examined XLID families, we selected 222 EUROMRX consortium families with convincing evidence for X-linkage, as evidenced by two or more affected males in two generations connected through healthy female carriers. In all, 97 of these 222 families had been resolved by mutation screening of single genes or by array CGH before this study (unpublished results).2 Of the remaining 125 families, 32 could be solved and 3 potentially solved by NGS-based sequencing of all X-chromosomal genes, but 90 remained unsolved, with half of them having four or more affected males in separate sibships. Assuming that we would have detected all previously identified defects by NGS, this indicates that mutations in coding regions of all presently known XLID genes account for 58% of the (EUROMRX) Fragile X-negative families (Supplementary Figure 9). Combined with Fragile X, which is seen in about 15% of XLID families, NGS and Fragile X testing allows a molecular diagnosis in 64% of all families with XLID (Supplementary Figure 10).
There are several explanations why about one-third of all XLID families cannot be solved by Fragile X testing combined with X-exome sequencing, (1) technical limitations because of poor enrichment and coverage that may account for a small number of the families, (2) non-coding variants or yet to be annotated regions of X-chromosome, (3) also, it is rather likely that at least some of the families might have autosomal ID instead of XLID, (4) the unique DNA missense variants with currently unknown causality are pathogenic, (5) at least some of the cases might be due to multigenic variations, or (6) deleterious variants are located in yet undiscovered regulatory elements. Although there is no reliable information about the proportion of disease-causing mutations located outside coding exons, their frequency may be considerable. A recent effort to annotate the non-coding sequence showed that around 80% of the genome contains elements linked to a biochemical function.120 Nowadays, whole genome sequencing or targeted genomic sequencing of linkage intervals combined with sophisticated computational tools that predict such potentially functionally relevant sequences, in principle, allow finding disease-relevant variants outside coding exons. One example is a family with non-syndromic XLID in which we failed to identify the causative mutation by exome sequencing. Subsequent massively parallel resequencing of the non-repetitive genomic linkage interval identified a regulatory variant that leads to overexpression of the transcriptional regulator HCFC1.7 Though the numbers of non-coding sequences in the human genome are comparably large, interpreting non-protein coding variants is a new challenge for the next years.
In conclusion, we have been able to identify numerous pathogenic variants in known XLID genes, previously proposed and novel XLID genes and two XLID candidates. The results provide a molecular diagnosis for the families involved and will be useful for interpreting variants that will be identified in other patients and families in these genes in the future. It will also help to better understand the genetic complexity underlying ID and the functional complexity underlying normal brain function, which is amazingly diverse. There is a growing body of evidence demonstrating that genetic lesions identified in XLID genes are also associated with other brain/neurological disorders, many of these often co-occurring with ID including autism, epilepsy, schizophrenia or other neuropsychiatric and neurobehavioral problems. Therefore, further investigations of the XLID genes in the context of their functional and regulatory networks will not only deepen our insight into the pathogenesis of ID but also shed more light into the etiology of related neurological disorders and into human brain development.
References
Ropers HH, Hamel BC . X-linked mental retardation. Nat Rev Genet 2005; 6: 46–57.
de Brouwer AP, Yntema HG, Kleefstra T, Lugtenberg D, Oudakker AR, de Vries BB et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum Mutat 2007; 28: 207–208.
Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009; 41: 535–543.
Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet 2010; 87: 173–188.
Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet 2010; 42: 486–488.
Homan CC, Kumar R, Nguyen LS, Haan E, Raymond FL, Abidi F et al. Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. Am J Hum Genet 2014; 94: 470–478.
Huang L, Jolly LA, Willis-Owen S, Gardner A, Kumar R, Douglas E et al. A noncoding, regulatory mutation implicates HCFC1 in nonsyndromic intellectual disability. Am J Hum Genet 2012; 91: 694–702.
Froyen G, Corbett M, Vandewalle J, Jarvela I, Lawrence O, Meldrum C et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am J Hum Genet 2008; 82: 432–443.
Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K et al. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet 2008; 82: 1003–1010.
Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008; 40: 776–781.
Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, D'Elia E et al. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet 2010; 86: 185–195.
Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012; 489: 313–317.
Yu HC, Sloan JL, Scharer G, Brebner A, Quintana AM, Achilly NP et al. An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am J Hum Genet 2013; 93: 506–514.
Starokadomskyy P, Gluck N, Li H, Chen B, Wallis M, Maine GN et al. CCDC22 deficiency in humans blunts activation of proinflammatory NF-kappaB signaling. J Clin Invest 2013; 123: 2244–2256.
Voineagu I, Huang L, Winden K, Lazaro M, Haan E, Nelson J et al. CCDC22: a novel candidate gene for syndromic X-linked intellectual disability. Mol Psychiatry 2012; 17: 4–7.
Johnston JJ, Gropman AL, Sapp JC, Teer JK, Martin JM, Liu CF et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 2012; 90: 295–300.
Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH et al. beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013; 136: 1708–1717.
Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet 2012; 90: 119–124.
Cacciagli P, Sutera-Sardo J, Borges-Correia A, Roux JC, Dorboz I, Desvignes JP et al. Mutations in BCAP31 cause a severe X-linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the Golgi apparatus. Am J Hum Genet 2013; 93: 579–586.
Hirata H, Nanda I, van Riesen A, McMichael G, Hu H, Hambrock M et al. ZC4H2 mutations are associated with arthrogryposis multiplex congenita and intellectual disability through impairment of central and peripheral synaptic plasticity. Am J Hum Genet 2013; 92: 681–695.
Van Maldergem L, Hou Q, Kalscheuer VM, Rio M, Doco-Fenzy M, Medeira A et al. Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Hum Mol Genet 2013; 22: 3306–3314.
Geetha TS, Michealraj KA, Kabra M, Kaur G, Juyal RC, Thelma BK . Targeted deep resequencing identifies MID2 mutation for X-linked intellectual disability with varied disease severity in a large kindred from India. Hum Mutat 2014; 35: 41–44.
Hu CH, Wrogemann K, Kalscheuer V, Tzschach A, Richard H, Haas SA et al. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J 2009; 3: 41–49.
Weese D, Emde AK, Rausch T, Doring A, Reinert K . RazerS—fast read mapping with sensitivity control. Genome Res 2009; 19: 1646–1654.
Emde AK, Schulz MH, Weese D, Sun R, Vingron M, Kalscheuer VM et al. Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics 2012; 28: 619–627.
Love MI, Mysickova A, Sun R, Kalscheuer V, Vingron M, Haas SA . Modeling read counts for CNV detection in exome sequencing data. Stat Appl Genet Mol Biol 2010; 10: 1544–6115.
Li H, Ruan J, Durbin R . Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008; 18: 1851–1858.
Hu H, Wienker TF, Musante L, Kalscheuer VM, Kahrizi K, Najmabadi H et al. Integrated sequence analysis pipeline provides one-stop solution for identifying disease-causing mutations. Hum Mutat 2014; 35: 1427–1435.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009; 25: 1966–1967.
Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 2010; 42: 969–972.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A . Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010; 20: 110–121.
Reese MG, Eeckman FH, Kulp D, Haussler D . Improved splice site detection in Genie. J Comput Biol 1997; 4: 311–323.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J . A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–315.
Friedrich T, Breiderhoff T, Jentsch TJ . Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem 1999; 274: 896–902.
Lorenz C, Pusch M, Jentsch TJ . Heteromultimeric CLC chloride channels with novel properties. Proc Natl Acad Sci USA 1996; 93: 13362–13366.
Kaech S, Banker G . Culturing hippocampal neurons. Nat Protoc 2006; 1: 2406–2415.
Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 2012; 7: 1741–1754.
Nadif Kasri N, Nakano-Kobayashi A, Malinow R, Li B, Van Aelst L . The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev 2009; 23: 1289–1302.
Rickheit G, Wartosch L, Schaffer S, Stobrawa SM, Novarino G, Weinert S et al. Role of ClC-5 in renal endocytosis is unique among ClC exchangers and does not require PY-motif-dependent ubiquitylation. J Biol Chem 2010; 285: 17595–17603.
Stauber T, Jentsch TJ . Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J Biol Chem 2010; 285: 34537–34548.
Piton A, Redin C, Mandel JL . XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet 2013; 93: 368–383.
Piton A, Poquet H, Redin C, Masurel A, Lauer J, Muller J et al. 20 ans apres: a second mutation in MAOA identified by targeted high-throughput sequencing in a family with altered behavior and cognition. Eur J Hum Genet 2014; 22: 776–783.
Lesca G, Moizard MP, Bussy G, Boggio D, Hu H, Haas SA et al. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am J Med Genet A 2013; 161A: 3063–3071.
Philips AK, Siren A, Avela K, Somer M, Peippo M, Ahvenainen M et al. X-exome sequencing in Finnish families with intellectual disability—four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis 2014; 9: 49.
Haddad DM, Vilain S, Vos M, Esposito G, Matta S, Kalscheuer VM et al. Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol Cell 2013; 50: 831–843.
Masurel-Paulet A, Kalscheuer VM, Lebrun N, Hu H, Levy F, Thauvin-Robinet C et al. Expanding the clinical phenotype of patients with a ZDHHC9 mutation. Am J Med Genet A 2014; 164A: 789–795.
Strobl-Wildemann G, Kalscheuer VM, Hu H, Wrogemann K, Ropers HH, Tzschach A . Novel GDI1 mutation in a large family with nonsyndromic X-linked intellectual disability. Am J Med Genet A 2011; 155A: 3067–3070.
Vulto-van Silfhout AT, Nakagawa T, Bahi-Buisson N, Haas SA, Hu H, Bienek M et al. Variants in CUL4B are associated with cerebral malformations. Hum Mutat 2014; 36: 106–117.
Meloni I, Muscettola M, Raynaud M, Longo I, Bruttini M, Moizard MP et al. FACL4, encoding fatty acid-CoA ligase 4, is mutated in nonspecific X-linked mental retardation. Nat Genet 2002; 30: 436–440.
Longo I, Frints SG, Fryns JP, Meloni I, Pescucci C, Ariani F et al. A third MRX family (MRX68) is the result of mutation in the long chain fatty acid-CoA ligase 4 (FACL4) gene: proposal of a rapid enzymatic assay for screening mentally retarded patients. J Med Genet 2003; 40: 11–17.
Yonath H, Marek-Yagel D, Resnik-Wolf H, Abu-Horvitz A, Baris HN, Shohat M et al. X inactivation testing for identifying a non-syndromic X-linked mental retardation gene. J Appl Genet 2011; 52: 437–441.
Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 2003; 34: 27–29.
Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet 2013; 93: 249–263.
Mikhailov A, Fennell A, Plong-on O, Sripo T, Hansakunachai T, Roongpraiwan R et al. Screening of NLGN3 and NLGN4X genes in Thai children with autism spectrum disorder. Psychiatr Genet 2014; 24: 42–43.
Schwartz CE, Tarpey PS, Lubs HA, Verloes A, May MM, Risheg H et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet 2007; 44: 472–477.
Risheg H, Graham JM Jr ., Clark RD, Rogers RC, Opitz JM, Moeschler JB et al. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet 2007; 39: 451–453.
Vulto-van Silfhout AT, de Vries BB, van Bon BW, Hoischen A, Ruiterkamp-Versteeg M, Gilissen C et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am J Hum Genet 2013; 92: 401–406.
Scheel O, Zdebik AA, Lourdel S, Jentsch TJ . Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 2005; 436: 424–427.
Claes S, Vogels A, Holvoet M, Devriendt K, Raeymaekers P, Cassiman JJ et al. Regional localization of two genes for nonspecific X-linked mental retardation to Xp22.3-p22.2 (MRX49) and Xp11.3-p11.21 (MRX50). Am J Med Genet 1997; 73: 474–479.
Raynaud M, Gendrot C, Dessay B, Moncla A, Ayrault AD, Moizard MP et al. X-linked mental retardation with neonatal hypotonia in a French family (MRX15): gene assignment to Xp11.22-Xp21.1. Am J Med Genet 1996; 64: 97–106.
Feng L, Campbell EB, Hsiung Y, MacKinnon R . Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 2010; 330: 635–641.
Estévez R, Pusch M, Ferrer-Costa C, Orozco M, Jentsch TJ . Functional and structural conservation of CBS domains from CLC chloride channels. J Physiol 2004; 557: 363–378.
Bykova EA, Zhang XD, Chen TY, Zheng J . Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat Struct Mol Biol 2006; 13: 1115–1119.
Wilson M, Mulley J, Gedeon A, Robinson H, Turner G . New X-linked syndrome of mental retardation, gynecomastia, and obesity is linked to DXS255. Am J Med Genet 1991; 40: 406–413.
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G . Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11: 548.
Besset V, Rhee K, Wolgemuth DJ . The cellular distribution and kinase activity of the Cdk family member Pctaire1 in the adult mouse brain and testis suggest functions in differentiation. Cell Growth Differ 1999; 10: 173–181.
Graeser R, Gannon J, Poon RY, Dubois T, Aitken A, Hunt T . Regulation of the CDK-related protein kinase PCTAIRE-1 and its possible role in neurite outgrowth in Neuro-2A cells. J Cell Sci 2002; 115: 3479–3490.
Fu WY, Cheng K, Fu AK, Ip NY . Cyclin-dependent kinase 5-dependent phosphorylation of Pctaire1 regulates dendrite development. Neuroscience 2011; 180: 353–359.
Liu Y, Cheng K, Gong K, Fu AK, Ip NY . Pctaire1 phosphorylates N-ethylmaleimide-sensitive fusion protein: implications in the regulation of its hexamerization and exocytosis. J Biol Chem 2006; 281: 9852–9858.
Cheng K, Li Z, Fu WY, Wang JH, Fu AK, Ip NY . Pctaire1 interacts with p35 and is a novel substrate for Cdk5/p35. J Biol Chem 2002; 277: 31988–31993.
Mokalled MH, Johnson A, Kim Y, Oh J, Olson EN . Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development 2010; 137: 2365–2374.
Ou CY, Poon VY, Maeder CI, Watanabe S, Lehrman EK, Fu AK et al. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 2010; 141: 846–858.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012; 485: 242–245.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet 2007; 80: 393–406.
Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478: 57–63.
Hellman-Aharony S, Smirin-Yosef P, Halevy A, Pasmanik-Chor M, Yeheskel A, Har-Zahav A et al. Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatr Neurol 2013; 49: 411–416, e411.
Piton A, Gauthier J, Hamdan FF, Lafreniere RG, Yang Y, Henrion E et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol Psychiatry 2011; 16: 867–880.
Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, MacArthur DG et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 2013; 77: 235–242.
Vandewalle J, Bauters M, Van Esch H, Belet S, Verbeeck J, Fieremans N et al. The mitochondrial solute carrier SLC25A5 at Xq24 is a novel candidate gene for non-syndromic intellectual disability. Hum Genet 2013; 132: 1177–1185.
Bedoyan JK, Schaibley VM, Peng W, Bai Y, Mondal K, Shetty AC et al. Disruption of RAB40AL function leads to Martin—Probst syndrome, a rare X-linked multisystem neurodevelopmental human disorder. J Med Genet 2012; 49: 332–340.
Lee J, Wong S, Boles RG . Mutation in the X-linked RAB40AL gene (Martin-Probst syndrome) with mental retardation, sensorineural hearing loss, and anomalies of the craniofacies and genitourinary tract: a second case report. Eur J Pediatr 2014; 173: 967–969.
Iqbal Z, Hu H, Haas SA, Shaw M, Lebrun N, Seemanova E et al. RAB40AL loss-of-function mutation does not cause X-linked intellectual disability. J Med Genet 2013; 49: 332.
Lehrke R . Theory of X-linkage of major intellectual traits. Am J Ment Defic 1972; 76: 611–619.
Lehrke RG . X-linked mental retardation and verbal disability. Birth Defects Orig Artic Ser 1974; 10: 1–100.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485: 237–241.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012; 74: 285–299.
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012; 335: 823–828.
Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013; 54: 1270–1281.
Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013; 12: 659–668.
Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001; 104: 205–215.
Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J 2005; 24: 1079–1091.
Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M et al. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res 2003; 18: 1740–1747.
Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 2001; 29: 185–196.
Poet M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci USA 2006; 103: 13854–13859.
Houge G, Rasmussen IH, Hovland R . Loss-of-function CNKSR2 mutation is a likely cause of non-syndromic X-linked intellectual disability. Mol Syndromol 2012; 2: 60–63.
Vaags AK, Bowdin S, Smith ML, Gilbert-Dussardier B, Brocke-Holmefjord KS, Sinopoli K et al. Absent CNKSR2 causes seizures and intellectual, attention, and language deficits. Ann Neurol 2014; 76: 758–764.
Stiffler MA, Grantcharova VP, Sevecka M, MacBeath G . Uncovering quantitative protein interaction networks for mouse PDZ domains using protein microarrays. J Am Chem Soc 2006; 128: 5913–5922.
Ohtakara K, Nishizawa M, Izawa I, Hata Y, Matsushima S, Taki W et al. Densin-180, a synaptic protein, links to PSD-95 through its direct interaction with MAGUIN-1. Genes Cells 2002; 7: 1149–1160.
Lim J, Ritt DA, Zhou M, Morrison DK . The CNK2 scaffold interacts with vilse and modulates Rac cycling during spine morphogenesis in hippocampal neurons. Curr Biol 2014; 24: 786–792.
Honda S, Orii KO, Kobayashi J, Hayashi S, Imamura A, Imoto I et al. Novel deletion at Xq24 including the UBE2A gene in a patient with X-linked mental retardation. J Hum Genet 2010; 55: 244–247.
Lee HW, Choi J, Shin H, Kim K, Yang J, Na M et al. Preso, a novel PSD-95-interacting FERM and PDZ domain protein that regulates dendritic spine morphogenesis. J Neurosci 2008; 28: 14546–14556.
Mignon-Ravix C, Cacciagli P, Choucair N, Popovici C, Missirian C, Milh M et al. Intragenic rearrangements in X-linked intellectual deficiency: Results of a-CGH in a series of 54 patients and identification of TRPC5 and KLHL15 as potential XLID genes. Am J Med Genet A 2014; 164A: 1991–1997.
Oberg EA, Nifoussi SK, Gingras AC, Strack S . Selective proteasomal degradation of the B'beta subunit of protein phosphatase 2A by the E3 ubiquitin ligase adaptor Kelch-like 15. J Biol Chem 2012; 287: 43378–43389.
Wylie CJ, Hendricks TJ, Zhang B, Wang L, Lu P, Leahy P et al. Distinct transcriptomes define rostral and caudal serotonin neurons. J Neurosci 2010; 30: 670–684.
Poeck B, Fischer S, Gunning D, Zipursky SL, Salecker I . Glial cells mediate target layer selection of retinal axons in the developing visual system of Drosophila. Neuron 2001; 29: 99–113.
Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol Cell 2008; 29: 92–101.
van Bokhoven H, Kramer JM . Disruption of the epigenetic code: an emerging mechanism in mental retardation. Neurobiol Dis 2010; 39: 3–12.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D . MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Bach I, Rodriguez-Esteban C, Carriere C, Bhushan A, Krones A, Rose DW et al. RLIM inhibits functional activity of LIM homeodomain transcription factors via recruitment of the histone deacetylase complex. Nat Genet 1999; 22: 394–399.
Ostendorff HP, Peirano RI, Peters MA, Schluter A, Bossenz M, Scheffner M et al. Ubiquitination-dependent cofactor exchange on LIM homeodomain transcription factors. Nature 2002; 416: 99–103.
Castle CD, Cassimere EK, Lee J, Denicourt C . Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol Cell Biol 2010; 30: 4404–4414.
Castle CD, Cassimere EK, Denicourt C . LAS1L interacts with the mammalian Rix1 complex to regulate ribosome biogenesis. Mol Biol Cell 2012; 23: 716–728.
Fanis P, Gillemans N, Aghajanirefah A, Pourfarzad F, Demmers J, Esteghamat F et al. Five friends of methylated chromatin target of protein-arginine-methyltransferase[prmt]-1 (chtop), a complex linking arginine methylation to desumoylation. Mol Cell Proteomics 2012; 11: 1263–1273.
Ouyang J, Shi Y, Valin A, Xuan Y, Gill G . Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol Cell 2009; 34: 145–154.
Butterfield RJ, Stevenson TJ, Xing L, Newcomb TM, Nelson B, Zeng W et al. Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurology 2014; 82: 1322–1330.
Harakalova M, van den Boogaard MJ, Sinke R, van Lieshout S, van Tuil MC, Duran K et al. X-exome sequencing identifies a HDAC8 variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J Med Genet 2012; 49: 539–543.
Ecker JR, Bickmore WA, Barroso I, Pritchard JK, Gilad Y, Segal E . Genomics: ENCODE explained. Nature 2012; 489: 52–55.
Scheel O, Zdebik A, Lourdel S, Jentsch TJ . Voltage-dependent electrogenic chloride proton exchange by endosomal CLC proteins. Nature 2005; 436: 424–427.
Yao I, Hata Y, Ide N, Hirao K, Deguchi M, Nishioka H et al. MAGUIN, a novel neuronal membrane-associated guanylate kinase-interacting protein. J Biol Chem 1999; 274: 11889–11896.
Mo J, Lee D, Hong S, Han S, Yeo H, Sun W et al. Preso regulation of dendritic outgrowth through PI(4,5)P2-dependent PDZ interaction with betaPix. Eur J Neurosci 2012; 36: 1960–1970.
Li SH, Li XJ . Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet 2004; 20: 146–154.
Shin BS, Kim JR, Walker SE, Dong J, Lorsch JR, Dever TE . Initiation factor eIF2gamma promotes eIF2-GTP-Met-tRNAi(Met) ternary complex binding to the 40S ribosome. Nat Struct Mol Biol 2011; 18: 1227–1234.
Dufu K, Livingstone MJ, Seebacher J, Gygi SP, Wilson SA, Reed R . ATP is required for interactions between UAP56 and two conserved mRNA export proteins, Aly and CIP29, to assemble the TREX complex. Genes Dev 2010; 24: 2043–2053.
Ma XM, Yoon SO, Richardson CJ, Julich K, Blenis J . SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 2008; 133: 303–313.
Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 2004; 13: 587–597.
Sowa ME, Bennett EJ, Gygi SP, Harper JW . Defining the human deubiquitinating enzyme interaction landscape. Cell 2009; 138: 389–403.
Klein U, Ramirez MT, Kobilka BK, von Zastrow M . A novel interaction between adrenergic receptors and the alpha-subunit of eukaryotic initiation factor 2B. J Biol Chem 1997; 272: 19099–19102.
Prezeau L, Richman JG, Edwards SW, Limbird LE . The zeta isoform of 14-3-3 proteins interacts with the third intracellular loop of different alpha2-adrenergic receptor subtypes. J Biol Chem 1999; 274: 13462–13469.
Furukawa M, He YJ, Borchers C, Xiong Y . Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol 2003; 5: 1001–1007.
Markson G, Kiel C, Hyde R, Brown S, Charalabous P, Bremm A et al. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res 2009; 19: 1905–1911.
Acknowledgements
We thank all the families who participated in this research. We also thank Susanne Freier, Ines Müller, Rien Blok, Carolien Oosterhoud, Roel Brandts, Demis Tserpelis, Evelyn Douglas, Lynne Hobson, Pia Winter and Sara Ekvall for excellent technical assistance; Dagmar Wieczorek, Vincent Desportes, Jean-Paul Bonnefont, Valerie Biancalana, Daniel Amram, Stanislas Lyonnet, Jacqueline Vigneron, Veronica Cusin, Philippe Jonveaux, Laurence-Olivier Faivre, Lydie Burglen, Yves Alembik, Sophie Scheidecker, Aurelia Jacquette, Delphine Heron, Cyril Goizet, Marie Ange Delrue, Didier Lacombe, Odile Boute, Andre Megarbane, Anne Moncla, Brigitte Gilbert Tiong Tan, Sharron Townshend, Michael Gabbett, Zornitza Stark, Mac Gardner, Joanne Dixon, Ian Glass, Martin Delatycki, Salim Aftimos, Felicity Collins, Paul James, George McGillivray, Kate Pope, Judith Allanson, Claude Moraine, Christine Francannet, Annick Toutain, James Lespinasse, Brigitte Gilbert, Isabelle Mortemousque, Anne Moncla, Etienne Mornet, Albert David, Connie Schrander-Stumpel, Christine de Die-Smulders, Yvonne Arens, Eric Smeets, Dominique Marcus-Soekarman, Ute Moog, Bert Smeets, Marzena Wiśniewska, Renata Glazar, Maciej Robert Krawczyński, Barbara Leube, Magdalena Nawara, Agnieszka Charzewska, Irma Järvelä, Barbara Oehl-Jaschkowitz, Marco Henneke, Peter Propping, Gholamali Tariverdian, Almuth Caliebe, Dorothea Wand, Jürgen Mücke, Eva Seemanova, Birgit Zirn, Jörg Müsebeck, Rudolf Mallmann, Christiane Zweier, Rami Abou Jamra and Anita Rauch for contributing families with XLID. We would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. This work was financed by a grant of the German Ministry of Education and Research through the MRNET (to HHR); the EU FP7 project GENCODYS, grant number 241995; the Max Planck Innovation Funds (to HHR), NWO VENI grant: 91666017 to SGMF, grant 2009(2)-81 from the Dutch Brain Foundation to AdB, WCH Foundation and Australian NHMRC to JG, grant from Fondation pour la Recherche Médicale (FRM, J Chelly-Equipe FRM 2013: DEQ2000326477).
Web Resources
1000 Genomes Project Consortium, http://www.1000genomes.org/data.
SplazerS, http://www.seqan.de/projects/splazers.html.
SnpStore, http://www.seqan.de/projects/snpstore.html.
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim.
PolyPhen2, http://genetics.bwh.harvard.edu/pph2/.
SIFT, http://sift.jcvi.org/.
NHLBI Exome Variant Server (ESP6500), http://evs.gs.washington.edu/EVS/.
dbSNP135, dbSNP138, http://www.ncbi.nlm.nih.gov/snp.
Ingenuity tool, http://www.ingenuity.com.
HOPE server, http://www.cmbi.ru.nl/hope/home.
shRNA designer tools, http://france.promega.com/resources/tools/.
Decipher, http://decipher.sanger.ac.uk/.
RNAi Consortium (TCR) shRNA Library, http://www.broadinstitute.org/rnai/trc/lib.
MutationTaster, http://www.mutationtaster.org/.
The Human Gene Mutation Database, http://www.hgmd.org/.
Exome Aggregation Consortium (ExAC), Cambridge, MA, http://exac.broadinstitute.org/.
[November, 2014 accessed].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hu, H., Haas, S., Chelly, J. et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry 21, 133–148 (2016). https://doi.org/10.1038/mp.2014.193
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2014.193
This article is cited by
-
Identification of a DLG3 stop mutation in the MRX20 family
European Journal of Human Genetics (2024)
-
Genetic analysis of a child with severe intellectual disability caused by a novel variant in the FERM domain of the FRMPD4 protein
Journal of Genetics (2024)
-
Genotype-phenotype correlation in CLCN4-related developmental and epileptic encephalopathy
Human Genetics (2024)
-
Sex differences in the intergenerational link between maternal and neonatal whole blood DNA methylation: a genome-wide analysis in 2 birth cohorts
Clinical Epigenetics (2023)
-
Whole exome sequencing revealed variants in four genes underlying X-linked intellectual disability in four Iranian families: novel deleterious variants and clinical features with the review of literature
BMC Medical Genomics (2023)
