
Abstract
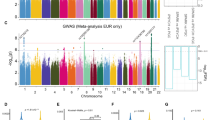
Autism is a highly heritable neurodevelopmental disorder, and known genetic variants, mostly rare, account only for a small proportion of cases. Here we report a genome-wide association study on autism using two Chinese cohorts as gene discovery (n=2150) and three data sets of European ancestry populations for replication analysis of top association signals. Meta-analysis identified three single-nucleotide polymorphisms, rs936938 (P=4.49 × 10−8), non-synonymous rs6537835 (P=3.26 × 10−8) and rs1877455 (P=8.70 × 10−8), and related haplotypes, AMPD1-NRAS-CSDE1, TRIM33 and TRIM33-BCAS2, associated with autism; all were mapped to a previously reported linkage region (1p13.2) with autism. These genetic associations were further supported by a cis-acting regulatory effect on the gene expressions of CSDE1, NRAS and TRIM33 and by differential expression of CSDE1 and TRIM33 in the human prefrontal cortex of post-mortem brains between subjects with and those without autism. Our study suggests TRIM33 and NRAS-CSDE1 as candidate genes for autism, and may provide a novel insight into the etiology of autism.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Bailey A, Phillips W, Rutter M . Autism: towards an integration of clinical, genetic, neuropsychological, and neurobiological perspectives. J Child Psychol Psychiatry 1996; 37: 89–126.
Blumberg S, Bramlett MD, Kogan MD, Schieve LA, Jones JR . Changes in prevalence of parent-reported autism spectrum disorder in school-aged U.S. children: 2007 to 2011–2012. Natl Health Statist Rep 2013; 65: 1–12.
Hallmayer J, Glasson EJ, Bower C, Petterson B, Croen L, Grether J et al. On the twin risk in autism. Am J Hum Genet 2002; 71: 941–946.
Veenstra-VanderWeele J, Cook EH Jr . Molecular genetics of autism spectrum disorder. Mol Psychiatry 2004; 9: 819–832.
Gupta AR, State MW . Recent advances in the genetics of autism. Biol Psychiatry 2007; 61: 429–437.
Veenstra-Vanderweele J, Christian SL, Cook EH Jr . Autism as a paradigmatic complex genetic disorder. Annu Rev Genom Hum Genet 2004; 5: 379–405.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007; 39: 25–27.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459: 569–573.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011; 70: 863–885.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T et al. Strong association of de novo copy number mutations with autism. Science 2007; 316: 445–449.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358: 667–675.
Weiss LA, Arking DE, Daly MJ, Chakravarti A . A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009; 461: 802–808.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Muers M . Human genetics: fruits of exome sequencing for autism. Nat Rev Genet 2012; 13: 377.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012; 485: 242–245.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485: 237–241.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009; 459: 528–533.
Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet 2010; 19: 4072–4082.
McClellan J, King MC . Genetic heterogeneity in human disease. Cell 2010; 141: 210–217.
Geschwind DH . Autism: many genes, common pathways? Cell 2008; 135: 391–395.
Howie BN, Donnelly P, Marchini J . A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB . Rare variants create synthetic genome-wide associations. PLoS Biol 2010; 8: e1000294.
Xu Z, Taylor JA . SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res 2009; 37: W600–W605.
Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011; 478: 519–523.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384.
Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L et al. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet 2012; 8: e1002592.
Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J et al. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet 1999; 65: 493–507.
Auranen M, Nieminen T, Majuri S, Vanhala R, Peltonen L, Järvelä I . Analysis of autism susceptibility gene loci on chromosomes 1p, 4p, 6q, 7q, 13q, 15q, 16p, 17q, 19q and 22q in Finnish multiplex families. Mol Psychiatry 2000; 5: 320–322.
Allen-Brady K, Miller J, Matsunami N, Stevens J, Block H, Farley M et al. A high-density SNP genome-wide linkage scan in a large autism extended pedigree. Mol Psychiatry 2009; 14: 590–600.
Kishino T, Lalande M, Wagstaff J . UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 1997; 15: 70–73.
Guernsey DL, Jiang H, Bedard K, Evans SC, Ferguson M, Matsuoka M et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot–Marie–Tooth disease. PLoS Genet 2010; 6: e1001081.
Abbas N, Lücking CB, Ricard S, Dürr A, Bonifati V, De Michele G et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet 1999; 8: 567–574.
Tarpey PS, Raymond FL, O'Meara S, Edkins S, Teague J, Butler A et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet 2007; 80: 345–352.
Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet 2007; 80: 561–566.
Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, Kahler SG et al. Autism and environmental genomics. Neurotoxicology 2006; 27: 671–684.
Acknowledgements
We are grateful to all the children with autism, their families and to the normal controls who participated in this study. We thank Autism Speaks for sharing resources from the Autism Genetic Resources Exchange (AGRE), the Simons Foundation for Autism Research Initiative (SFARI) for providing data from the Simons Simplex Collection (SCC), the NIH GWAS Data Repository (AGP data set: phs000267.v1.p1) and the Contributing Investigator(s) who contributed the phenotype and genotype data from his/her original studies. We also thank Mr Tianzhang Ye, Dr Carlo Colantuoni and Dr Joel E Kleinman for assisting in accessing the brain expression data and Dr Elizabeth Sherman for comments. The research was supported by the National Basic Research Program of China (2012CB517900, 2011CB510002), the National Natural Science Foundation of China (81330027, 81161120544), the National Alliance for Research on Schizophrenia and Depression (NARSAD) Award (17616 to LZ) and Intramural Research Program funding from the National Institute of Mental Health, The National Institutes of Health in the United States.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
PowerPoint slides
Rights and permissions
About this article
Cite this article
Xia, K., Guo, H., Hu, Z. et al. Common genetic variants on 1p13.2 associate with risk of autism. Mol Psychiatry 19, 1212–1219 (2014). https://doi.org/10.1038/mp.2013.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2013.146
Keywords
This article is cited by
-
Implications of Genetic Factors and Modifiers in Autism Spectrum Disorders: a Systematic Review
Review Journal of Autism and Developmental Disorders (2024)
-
A novel 1p13.2 deletion associates with neurodevelopmental disorders in a three-generation pedigree
BMC Medical Genomics (2023)
-
A human leukocyte antigen imputation study uncovers possible genetic interplay between gut inflammatory processes and autism spectrum disorders
Translational Psychiatry (2023)
-
Breast carcinoma-amplified sequence 2 regulates adult neurogenesis via β-catenin
Stem Cell Research & Therapy (2022)
-
Autism spectrum disorder and severe social impairment associated with elevated plasma interleukin-8
Pediatric Research (2021)
