
Abstract
Clear cell renal cell carcinoma is by far the most common form of kidney cancer; however, a number of histologically similar tumors are now recognized and considered distinct entities. The Cancer Genome Atlas published data set was queried (http://cbioportal.org) for clear cell renal cell carcinoma tumors lacking VHL gene mutation and chromosome 3p loss, for which whole-slide images were reviewed. Of the 418 tumors in the published Cancer Genome Atlas clear cell renal cell carcinoma database, 387 had VHL mutation, copy number loss for chromosome 3p, or both (93%). Of the remaining, 27/31 had whole-slide images for review. One had 3p loss based on karyotype but not sequencing, and three demonstrated VHL promoter hypermethylation. Nine could be reclassified as distinct or emerging entities: translocation renal cell carcinoma (n=3), TCEB1 mutant renal cell carcinoma (n=3), papillary renal cell carcinoma (n=2), and clear cell papillary renal cell carcinoma (n=1). Of the remaining, 6 had other clear cell renal cell carcinoma-associated gene alterations (PBRM1, SMARCA4, BAP1, SETD2), leaving 11 specimens, including 2 high-grade or sarcomatoid renal cell carcinomas and 2 with prominent fibromuscular stroma (not TCEB1 mutant). One of the remaining tumors exhibited gain of chromosome 7 but lacked histological features of papillary renal cell carcinoma. Two tumors previously reported to harbor TFE3 gene fusions also exhibited VHL mutation, chromosome 3p loss, and morphology indistinguishable from clear cell renal cell carcinoma, the significance of which is uncertain. In summary, almost all clear cell renal cell carcinomas harbor VHL mutation, 3p copy number loss, or both. Of tumors with clear cell histology that lack these alterations, a subset can now be reclassified as other entities. Further study will determine whether additional entities exist, based on distinct genetic pathways that may have implications for treatment.
Similar content being viewed by others
Main
Clear cell renal cell carcinoma is the most common form of adult renal cancer,1 which has been shown in both the hereditary and sporadic patient populations to be associated with abnormalities of the VHL gene and deletions of chromosome arm 3p25.2 Recently, understanding of the genetics of clear cell renal cell carcinoma has been further enhanced by genomic analysis, such as via The Cancer Genome Atlas Research Network and other large studies,2, 3 emphasizing the role of the several other genes, particularly SMARCA4, PBRM1, BAP1, and SETD2.3 Conversely however, other tumors that closely resemble clear cell renal cell carcinoma histologically are now regarded as unique tumor entities, based on differing histological, immunohistochemical, and molecular characteristics, such as exemplified by translocation renal cell carcinoma and clear cell papillary renal cell carcinoma.4, 5 It remains to be fully understood, however, whether there are additional renal tumor entities with ‘clear cell’ histology that warrant classification as unique tumor types with implications for prognosis or targeted treatment.6, 7
Materials and methods
Samples were retrieved from the previously reported Cancer Genome Atlas study on clear cell renal cell carcinoma that was institutional review board-approved at each participating site.3 The Cancer Genome Atlas data set for clear cell renal cell carcinoma3 was queried for alterations of 3p and the VHL gene via http://cbioportal.org, as previously described.8, 9 Whole-slide images were retrieved from the Cancer Digital Slide Archive (http://cancer.digitalslidearchive.net/) and clinical and pathological information was retrieved from the publically available data set10 and http://cbioportal.org.8, 9 Original pathology reports that were available were reviewed for diagnosis, comments, immunohistochemical studies, and cytogenetic information, when available. The available sequence data were also analyzed for the presence of gene fusions, using methods previously described.11 Methylation call data for these samples for the VHL promoter region were compiled from the Cancer Genome Atlas data, which were generated through the Infinium HumanMethylation450 BeadChip assay.3
Results
A total of 418 tumors were retrieved from the published Cancer Genome Atlas clear cell renal cell carcinoma dataset that contained both sequencing and copy number data. Of these, 224 were flagged as having VHL mutation or deep deletion. An additional 143 were found to have a 3p arm copy number loss without detection of VHL mutation or deep deletion. Another 20 were found to have decreased copy number for the entire chromosome 3, interpreted as monosomy 3. In total, 387 exhibited VHL mutation, copy number loss for chromosome 3p, or both (93%). Of the remaining 31 tumors, 27 had whole-slide images for review and formed the study cohort. Of these, methylation data were available for 15 of the 27 tumors, of which 5 had only tumor tissue (no normal tissue comparison). From these, 4 of the 15 were noted to have methylation at the VHL promoter site, ranging from 1 to 3 sites.3 Unique histological characteristics of the study cohort are summarized as follows:
Translocation Renal Cell Carcinoma, n=3
Three of the tumors with intact VHL and 3p25 were recently reported by Malouf et al12 to be translocation renal cell carcinomas. All three of these tumors had suggestive histology of a translocation-associated renal cell carcinoma, including large nests with calcifications, eosinophilic cells, papillae, and sclerotic stroma (Figures 1a and b). For one of the three tumors, a karyotype was available as part of the original pathology report, confirming t(X;1)(p11.2;p34). A fourth tumor lacking VHL alteration or 3p loss showed features reminiscent of translocation renal cell carcinoma by morphological review, including hyaline stroma, cells with voluminous cytoplasm, and well-defined cell borders. However, this was not detected to harbor a translocation in the study by Malouf et al12 or in our own sequence analysis. Interestingly, two tumors reported to have SFPQ-TFE3 translocation in the same study12 also exhibited VHL mutation, copy number loss for chromosome 3p, and morphology indistinguishable from clear cell renal cell carcinoma. (These were not included in the study denominator of 27 specimens, as they were positive for VHL alteration and copy number 3p loss.) Of these, 1 was also predicted to harbor SFPQ-TFE3 fusion in our own analysis, whereas the other was not predicted using our fusion detection analysis.11 One of these tumors with papillary morphology also had copy number gain for chromosome 17.

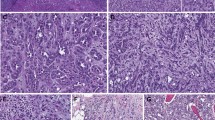
Tumors reclassified based on integration of morphology and genetics; whole-slide images from http://cancer.digitalslidearchive.net/: (a) TFE3 translocation-associated renal cell carcinoma, composed of cells with clear cytoplasm arranged in large nests with hyalized stroma. (b) TFE3 translocation-associated renal cell carcinoma with papillae, foamy macrophages, and psammoma bodies. (c) Papillary renal cell carcinoma with compact architecture, clear cytoplasmic changes, and admixed foamy macrophages. (d) Papillary renal cell carcinoma with well-formed papillae. By report, a clear cell renal cell carcinoma component was present in this patient, but this was not visualized in the available tumor slide images. (e, f) Clear cell papillary renal cell carcinoma, composed of branched glandular structures and broad papillae.
Papillary Renal Cell Carcinoma, n=2
Two tumors had morphology favoring papillary rather than clear cell renal cell carcinoma (Figures 1c and d). Of these, 1 tumor (10 cm, pT2) had florid cytoplasmic vacuolization and copy number gain for chromosomes 7 and 17, supporting a papillary renal cell carcinoma with cytoplasmic clearing mimicking clear cell renal cell carcinoma.13, 14 The second tumor was reported to be a mixture of 50% clear cell renal cell carcinoma and 50% papillary renal cell carcinoma, although the available whole-slide image demonstrated an entirely papillary architecture, consistent with papillary renal cell carcinoma. Therefore, it is possible that either sampling differences (due to two juxtaposed tumors) or areas of clear cell change within a papillary renal cell carcinoma tumor was confounding in this sample, causing the genetic data to reflect the typical pattern of papillary renal cell carcinoma.
Clear Cell Papillary Renal Cell Carcinoma, n=1
Another tumor was composed of both solid and cystic components, fibrous stroma, and uniform low-grade nuclei, together in support of clear cell papillary renal cell carcinoma15, 16, 17, 18 (Figures 1e and f). This patient was reported to have multiple tumors, all ≤3 cm. As such, this likely represents an example of multifocal clear cell papillary renal cell carcinoma, originally diagnosed as clear cell renal cell carcinoma, the most likely source of histological overlap.18 Interestingly, this tumor was noted to have one of the three sites demonstrating promoter methylation for the VHL gene. To date, clear cell papillary renal cell carcinomas are typically regarded as having no VHL alterations, with rare exceptions in which classification is debatable19, 20, 21, 22, 23, 24; however, few have been studied for promoter methylation.17
TCEB1 Mutant Renal Cell Carcinoma, n=3
Three tumors harbored TCEB1 mutation, the features of which have recently been published by Hakimi et al.6 Morphologically, these tumors have been noted to contain thick fibrous bands, a clear cell-like histological pattern, and voluminous cytoplasm (Figures 2a and b).6

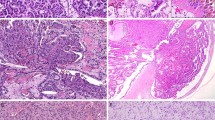
Tumors with fibromuscular stroma; whole-slide images from http://cancer.digitalslidearchive.net/: (a, b) TCEB1 mutant renal cell carcinoma with fibromuscular bands traversing the tumor, composed of cells with clear cytoplasm. (c) A non-TCEB1 mutant tumor with fibromuscular stroma, showing small vein invasion (d).
Other Histologies
After reclassification of the above 9 histological patterns, likely corresponding to distinct tumor entities, 18 tumors remained to be further investigated. Of these, distinct morphological categories were noted, as follows:
Renal cell carcinoma with fibromuscular stroma, n=5
Five tumors exhibited prominent fibromuscular stroma (Figures 2c and d). Three of these tumors were high-stage tumors (pT3), including 2 with metastasis and 1 with lymphovascular space invasion. Although Hakimi et al6 recently reported that TCEB1 mutant renal cell carcinomas to have prominent fibromuscular stroma, this group of five tumors had a prominent fibromuscular component without TCEB1 mutation. Methylation data were available for four of these five tumors, of which three tumors showed no VHL promoter hypermethylation. One tumor demonstrated two of the three sites with promoter hypermethylation for VHL, which also harbored a SETD2 frameshift mutation.3
High-grade renal cell carcinoma, n=4
Four tumors were high-grade renal cell carcinomas with rhabdoid or sarcomatoid morphology (Figure 3). One of these tumors was found to harbor a TP53 mutation and, as reported, was almost entirely sarcomatoid. Although TP53 mutations have been reported in clear cell renal cell carcinoma,2 this alteration is also common in chromophobe renal cell carcinoma,25 which despite its generally less aggressive behavior has a paradoxical predilection for sarcomatoid dedifferentiation.26, 27, 28 Aberration in RB1 was also detected, which has been reported in chromophobe renal cell carcinoma.29 Therefore, it is possible that this tumor represents chromophobe renal cell carcinoma in which the original histology is obscured by sarcomatoid features. A second tumor showed a SETD2 frameshift mutation, which is now recognized to be associated with clear cell renal cell carcinoma, although in the absence of VHL mutation and 3p deletion, it is uncertain whether this represents an alternative pathway to sarcomatoid renal cell carcinoma.28 Methylation data were available for two of the four tumors, neither of which demonstrated VHL promoter hypermethylation.3

High-grade renal cell carcinomas; whole-slide images from http://cancer.digitalslidearchive.net/: (a) High-grade, sarcomatoid renal cell carcinoma with little recognizable low-grade component. This tumor harbored TP53 and RB1 mutations. (b) Sarcomatoid renal cell carcinoma composed of spindle-shaped cells with tumor necrosis and other areas of rhabdoid cells (c). (d) High-grade renal cell carcinoma composed of both clear and amphophilic cells.
Chromosome 3p loss present by karyotyping, n=1
One tumor had an accompanying karyotype via conventional cytogenetic karyotyping, in which chromosome 3p deletion was noted in the original cytogenetic report. This tumor also harbored BAP1 mutation and had one of the three sites with VHL promoter hypermethylation.
VHL promoter hypermethylation, n=1
An additional single tumor not discussed in any of the other categories exhibited widespread nuclear alignment, reminiscent of clear cell papillary renal cell carcinoma (Figure 4a),23 and was noted to have three sites of VHL promoter hypermethylation, in conjunction with MTOR mutation.

Renal cell carcinomas without a unifying pattern; whole-slide images from http://cancer.digitalslidearchive.net/: (a) Diffuse nuclear alignment and branched glandular configuration, reminiscent of clear cell papillary renal cell carcinoma. This tumor exhibited VHL promoter hypermethylation and MTOR mutation. (b) Renal cell carcinoma with papillary or pseudopapillary structures and granular cytoplasm (SMARCA4 mutant). (c) Renal cell carcinoma with papillary or pseudopapillary areas containing clear cytoplasm and areas of spindle-shaped cell morphology (d) in the same tumor (PBRM1 mutant). (e) Renal cell carcinoma with clear cytoplasm, numerous intranuclear cytoplasmic invaginations, and other areas of syncytial-type giant tumor cells (f) in the same tumor. This tumor also had low-grade areas that would otherwise be typical for clear cell renal cell carcinoma but did not exhibit alterations in clear cell renal cell carcinoma-associated genes or VHL promoter hypermethylation.
Tumors not reclassified
There remained in total 18 tumors not specifically reclassified (Figure 4) (as papillary renal cell carcinoma, TCEB1 mutant renal cell carcinoma, translocation renal cell carcinoma, or clear cell papillary renal cell carcinoma, including the 1 tumor for which the possibility of sarcomatoid chromophobe renal cell carcinoma was considered but not fully resolvable). Of these, 11 lacked a clear cell renal cell carcinoma-associated alteration (VHL promoter hypermethylation or PBRM1, SETD2, SMARCA4, or BAP1 alteration). From these, three were included in the fibromuscular stroma category, and three were included in the high-grade category (including the one tumor with consideration of sarcomatoid chromophobe renal cell carcinoma). The remaining five tumors otherwise demonstrated histological features most in line with clear cell renal cell carcinoma and did not exhibit any specific histological patterns that would argue for a different diagnosis. One tumor resembling clear cell renal cell carcinoma had copy number increase for chromosome 7, suggesting trisomy 7; however, specific histological features of papillary renal cell carcinoma were not appreciable and immunohistochemical staining was not available. Of note, three additional tumors also had a similar copy number gain for chromosome 7, of which one had concurrent SETD2 frameshift mutation and was included in the high-grade renal cell carcinoma category, another had SETD2 frameshift mutation and was included in the fibromuscular stroma category, and the third was also included in the fibromuscular stroma category, with no other specific associated alterations. Other unusual findings in this group included foamy rather than empty-appearing cytoplasm (n=1), intranuclear cytoplasmic invaginations (n=1), and hyalinized stroma (n=1); however, based on current morphological criteria these remained most in line with a diagnosis of clear cell renal cell carcinoma. Immunohistochemical results were not available on any of these five tumors, and in only one a conventional karyotype had been attempted but was unsuccessful.
Discussion
In this study, we sought to examine the histology of renal ‘clear cell’ tumors lacking the usual genetic characteristics of VHL and chromosome 3p abnormalities and attempted to assess whether as yet undefined subtypes of clear cell renal cell tumors exist. Existing data indicate that a majority of clear cell renal cell carcinoma tumors harbors chromosome 3p aberration and/or VHL mutation.2, 30, 31, 32 However, it is also increasingly recognized that renal tumors are histologically and genetically complex, with a growing number of distinct tumor subtypes now recognized.33, 34 As prototypical examples of this concept, translocation renal cell carcinomas, which are likely to have been often been diagnosed as clear cell renal cell carcinoma in the past, and the more recently recognized clear cell papillary renal cell carcinoma are now regarded as distinct tumor types with potential implications for patient management. Other emerging renal cell carcinoma subtypes, such as TCEB1 mutant renal cell carcinoma6 and renal cell carcinoma with angioleiomyoma-like stroma (or renal angiomyoadenomatous tumor, among other names),7, 35, 36, 37, 38, 39 illustrate that there may yet be other tumors historically considered clear cell renal cell carcinoma that are genetically and possibly prognostically distinct.
We found that, after critical histological review of tumors without chromosome 3p or VHL mutation, at least 9 of the 27 (33%) can likely be reclassified as distinct tumor histologies. These include three translocation renal cell carcinomas (as previously reported by Malouf et al12), three TCEB1 mutant renal cell carcinomas (as recently characterized by Hakimi et al6), two papillary renal cell carcinomas,13, 14 and one clear cell papillary renal cell carcinoma.18, 40, 41, 42 Another 6 (22%) harbored additional clear cell renal cell carcinoma-associated gene alterations, including PBRM1, SETD2, SMARCA4, and BAP1. One of these was originally analyzed with conventional karyotype, which did detect 3p deletion, despite the absence of detected copy number change in the sequencing data. An additional tumor had VHL promoter hypermethylation, leaving only 11 tumors (2.6% of the original cohort of 418 tumors) that lacked clear cell renal cell carcinoma-associated genetic alterations and were not reclassifiable (4 tumors excluded, which had inadequate whole-slide images).
These results are strikingly similar to those of another recent study by Vang et al43 evaluating high-grade ovarian serous carcinoma. The authors performed a histological review of all TP53 wild-type high-grade ovarian serous carcinoma from the Cancer Genome Atlas ovarian cancer specimens,44 from which 96% of tumors harbored a TP53 genetic alteration. Of the TP53 wild-type tumors, alternative classifications were favored by at least a subset of the gynecological pathologists in essentially all cases, with exception of a rare tumor in which evolution from low-grade serous carcinoma was suspected.43 This analysis led the authors to conclude that ‘... all de novo ovarian [high grade serous carcinomas] contain alterations of TP53...’.43
One feature highlighted by this study is the presence of prominent fibromuscular stroma in renal cell tumors. Although Hakimi et al6 recently characterized a subtly unique histology of renal cell carcinoma with TCEB1 mutation (mutually exclusive with VHL and 3p alteration) as containing thick fibromuscular bands, we also noted that 5 additional tumors from the 27 studied in this cohort exhibited prominent fibromuscular stroma in the absence of TCEB1 mutation. These also included aggressive histological and staging characteristics, findings that appear to be lacking in the few reported TCEB1 mutant tumors thus far, which are presently hypothesized to have a favorable prognosis.6 One of these tumors also was noted to have VHL promoter hypermethylation and SETD2 frameshift mutation, likely supporting overall classification as a high-grade clear cell renal cell carcinoma. Similarly, in another recent study, almost half of renal cell carcinoma tumors originally considered renal cell carcinoma with angioleiomyoma-like stroma were ultimately excluded from the cohort after combined histological, immunohistochemical, and chromosomal analysis.7 Therefore, despite the novel recognition of TCEB1 mutation in renal cell tumors, this appears not to be the only pathway to prominent fibromuscular stroma in renal cell carcinoma, some of which may be part of the spectrum of clear cell papillary renal cell carcinoma and others of which likely include unusual fibromuscular-appearing patterns of aggressive sarcomatoid renal cell carcinoma and reactive fibromuscular stroma from invasion of structures.7
An additional four tumors in this cohort were high-grade or sarcomatoid renal cell carcinomas. In general, ‘driver’ alterations are often preserved in both the epithelial and sarcomatoid areas of sarcomatoid renal cell carcinoma. For example, Malouf et al28 recently showed that VHL mutations are usually preserved in both the clear cell component and sarcomatoid component of the tumor.28 Secondary clear cell renal cell carcinoma-associated mutations (for example BAP1, PBRM1, SETD2, SMARCD1, etc.) can also present in this setting.28 In the four tumors that appeared to lack VHL and 3p alteration in this cohort, it remains to be better understood whether there are alternative pathways that progress directly to a more aggressive sarcomatoid phenotype rather than via evolution from usual low-grade renal cell carcinoma over time. For example, one tumor of these four demonstrated SETD2 frameshift mutation, despite absence of VHL alteration (although methylation data were not available for this specimen). This raises the question as to whether there are alternative pathways to high-grade sarcomatoid tumors that do not proceed through the same steps as usual clear cell renal cell carcinoma.45 Alternatively, is it possible that the original alterations may be obscured by genomic instability or that detection may be influenced by variation across testing techniques. One other example from these four harbored TP53 and RB alterations with a completely sarcomatoid morphology in the available whole-slide image. Although these genetic alterations are not regarded as specific for a particular renal cell carcinoma histology, both are also known to occur in chromophobe renal cell carcinoma.25, 29 Interestingly, despite the usually favorable prognosis of chromophobe renal cell carcinoma, this histology appears to have a paradoxical predilection for sarcomatoid dedifferentiation,26, 27, 28 perhaps related to the enrichment of TP53 alteration. Therefore, it is possible that some extensively sarcomatoid renal cell carcinomas have actually evolved from chromophobe renal cell carcinoma and that the chromophobe component is not well visualized owing to sampling error, despite that clear cell renal cell carcinoma is much more common and may be the presumptive origin in diagnostic practice.
An additional interesting finding in this study is that two tumors previously characterized as harboring an SFPQ–TFE3 gene fusion by Malouf et al12 had morphology indistinguishable from clear cell renal cell carcinoma, in combination with VHL mutation and copy number decrease for chromosome arm 3p. This is counterintuitive, as one might assume that these alterations are mutually exclusive. However, other unusual combinations of distinct presumptive ‘driver’ alterations have also been recently reported, such as in a recent study of tumors with both TFE3 translocation and SDHB mutation.46 In this context, it remains to be better understood whether one alteration is the primary driver of tumorigenesis and the other is a secondary event due to genomic instability.11
A limitation of our study is that only representative whole-slide images, typically from a single histological slide, were available for review. In practice, we would typically regard a tumor as clear cell renal cell carcinoma if low-grade areas with prototypical morphology are present, even if other areas exhibit high-grade, heterogeneous, or unusual morphology.47 Therefore, owing to sampling error, low-grade areas with a classic appearance may have been missed in a subset of the cases with negative genetics. However, the striking predilection for VHL and 3p alterations suggests that alternative molecular mechanisms and classification possibilities should be considered in tumors considered ‘clear cell renal cell carcinoma’ lacking these findings, which might then have implications for the efficacy of usual clear cell renal cell carcinoma-targeted therapies.
Additionally, ancillary techniques, such as immunohistochemistry (n=4) and conventional karyotyping (n=3), were available only in a small subset of cases. Notably, however, each of the three tumors karyotyped had a significant finding, including one in which 3p deletion was detected by karyotype but not by copy number analysis. Therefore, owing to limitations and variations between testing methodology (karyotype, FISH, sequencing, methylation analysis, and others), an even larger number of the genetically ‘negative’ tumors may have harbored a chromosomal alteration if karyotyped, which would even further reduce the percentage of 3p intact clear cell renal cell carcinoma tumors. As an illustration of these potential differences between techniques, only 20 tumors lacking VHL mutation or deep deletion appeared to have copy number loss from the entire chromosome 3 based on our review of http://cbioportal.org data, whereas other studies using cytogenetic karyotyping have suggested that monosomy 3 may be a relatively common mechanism of chromosome 3 loss in clear cell renal cell carcinoma.48 Conversely, although 3p arm loss is a hallmark of clear cell renal cell carcinoma, it may not be entirely specific for clear cell renal cell carcinoma, as it has been reported in papillary renal cell carcinoma with clear cell change and the recently described renal cell carcinomas with TFEB amplification.11, 13
Conclusions
The vast majority of clear cell renal cell carcinomas harbor VHL mutation, 3p copy number loss, or both, suggesting that these may be defining abnormalities. Of tumors with clear cell histology that lack these alterations, a significant fraction can likely be reclassified as established and emerging entities. The significance of concurrent gene fusion (such as TFE3 rearrangement) and VHL mutation or 3p arm deletion in the same tumor remains to be better understood. The remaining tumors suggest that a ‘clear cell’ appearance may be a common phenotype of several genetic pathways, for which improved understanding may define new entities and guide prognostication and treatment.
References
Montironi R, Cheng L, Scarpelli M et al. Pathology and genetics: tumours of the urinary system and male genital system: clinical implications of the 4th edition of the WHO classification and beyond. Eur Urol 2016;70:120–123.
Sato Y, Yoshizato T, Shiraishi Y et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 2013;45:860–867.
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499:43–49.
Srigley JR, Cheng L, Grignon DJ et alClear cell papillary renal cell carcinoma. In: Moch H, Humphrey PA, Ulbright TM et al(eds). WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th edn International Agency for Research on Cancer: Lyon, France, 2016, pp 40–41.
Argani P, Cheville J, Ladanyi M . MiT family translocation renal cell carcinoma. In: Moch H, Humphrey PA, Ulbright TM et al(eds). WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th edn. International Agency for Research on Cancer: Lyon, France, 2016, pp 33–34.
Hakimi AA, Tickoo SK, Jacobsen A et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol 2015;28:845–853.
Williamson SR, Cheng L, Eble JN et al. Renal cell carcinoma with angioleiomyoma-like stroma: clinicopathological, immunohistochemical, and molecular features supporting classification as a distinct entity. Mod Pathol 2015;28:279–294.
Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6, pl1.
Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404.
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 2016;374:135–145.
Williamson SR, Grignon DJ, Cheng L et al. Renal cell carcinoma with chromosome 6p amplification including the TFEB gene: a novel mechanism of tumor pathogenesis? Am J Surg Pathol 2017;41:287–298.
Malouf GG, Su X, Yao H et al. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clin Cancer Res 2014;20:4129–4140.
Klatte T, Said JW, Seligson DB et al. Pathological, immunohistochemical and cytogenetic features of papillary renal cell carcinoma with clear cell features. J Urol 2011;185:30–35.
Gobbo S, Eble JN, Maclennan GT et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 2008;32:1780–1786.
Gobbo S, Eble JN, Grignon DJ et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008;32:1239–1245.
Tickoo SK, de Peralta-Venturina MN, Harik LR et al. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 2006;30:141–153.
Aydin H, Chen L, Cheng L et al. Clear cell tubulopapillary renal cell carcinoma: a study of 36 distinctive low-grade epithelial tumors of the kidney. Am J Surg Pathol 2010;34:1608–1621.
Williamson SR, Eble JN, Cheng L et al. Clear cell papillary renal cell carcinoma: differential diagnosis and extended immunohistochemical profile. Mod Pathol 2013;26:697–708.
Williamson SR, Zhang S, Eble JN et al. Clear cell papillary renal cell carcinoma-like tumors in patients with von Hippel-Lindau disease are unrelated to sproadic clear cell papillary renal cell carcinoma. Am J Surg Path 2013;37:1131–1139.
Rao P, Monzon F, Jonasch E et al. Clear cell papillary renal cell carcinoma in patients with von Hippel-Lindau syndrome-clinicopathological features and comparative genomic analysis of 3 cases. Hum Pathol 2014;45:1966–1972.
Williamson SR, Cheng L . Do clear cell papillary renal cell carcinomas occur in patients with von Hippel-Lindau disease? Hum Pathol 2015;46:340–341.
Rao P, Tamboli P . Do clear cell papillary renal cell carcinomas occur in patients with von Hippel-Lindau disease?-Reply. Hum Pathol 2015;46:341–343.
Williamson SR, Gupta NS, Eble JN et al. Clear cell renal cell carcinoma with borderline features of clear cell papillary renal cell carcinoma: combined morphologic, immunohistochemical, and cytogenetic analysis. Am J Surg Pathol 2015;39:1502–1510.
Williamson SR, Cheng L . Clear cell renal cell tumors: not all that is "clear" is cancer. Urol Oncol 2016;34:e17–e22.
Durinck S, Stawiski EW, Pavia-Jimenez A et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 2015;47:13–21.
de Peralta-Venturina M, Moch H, Amin M et al. Sarcomatoid differentiation in renal cell carcinoma: a study of 101 cases. Am J Surg Pathol 2001;25:275–284.
Akhtar M, Tulbah A, Kardar AH et al. Sarcomatoid renal cell carcinoma: the chromophobe connection. Am J Surg Pathol 1997;21:1188–1195.
Malouf GG, Ali SM, Wang K et al. Genomic characterization of renal cell carcinoma with sarcomatoid dedifferentiation pinpoints recurrent genomic alterations. Eur Urol 2016;70:348–357.
Davis CF, Ricketts CJ, Wang M et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014;26:319–330.
Moore LE, Jaeger E, Nickerson ML et al. Genomic copy number alterations in clear cell renal carcinoma: associations with case characteristics and mechanisms of VHL gene inactivation. Oncogenesis 2012;1:e14.
Hakimi AA, Ostrovnaya I, Reva B et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 2013;19:3259–3267.
Gossage L, Eisen T . Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat Rev Clin Oncol 2010;7:277–288.
Srigley JR, Delahunt B, Eble JN et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol 2013;37:1469–1489.
Moch H, Amin M, Argani P et alRenal cell tumors. In: Moch H, Humphrey P, Ulbright T et al(eds). WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th edn. International Agency for Research on Cancer: Lyon, France, 2016, pp 14–17.
Shannon BA, Cohen RJ, Segal A, Baker EG et al. Clear cell renal cell carcinoma with smooth muscle stroma. Hum Pathol 2009;40:425–429.
Petersson F, Branzovsky J, Martinek P et al. The leiomyomatous stroma in renal cell carcinomas is polyclonal and not part of the neoplastic process. Virchows Arch 2014;465:89–96.
Michal M, Hes O, Nemcova J et al. Renal angiomyoadenomatous tumor: morphologic, immunohistochemical, and molecular genetic study of a distinct entity. Virchows Arch 2009;454:89–99.
Michal M, Hes O, Havlicek F . Benign renal angiomyoadenomatous tumor: a previously unreported renaltumor. Ann Diagn Pathol 2000;4:311–315.
Kuhn E, De Anda J, Manoni S et al. Renal cell carcinoma associated with prominent angioleiomyoma-like proliferation: report of 5 cases and review of the literature. Am J Surg Pathol 2006;30:1372–1381.
Zhou H, Zheng S, Truong LD et al. Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum Pathol 2014;45:59–64.
Diolombi ML, Cheng L, Argani P et al. Do clear cell papillary renal cell carcinomas have malignant potential? Am J Surg Pathol 2015;39:1621–1634.
Aron M, Chang E, Herrera L et al. Clear cell-papillary renal cell carcinoma of the kidney not associated with end-stage renal disease: clinicopathologic correlation with expanded immunophenotypic and molecular characterization of a large cohort with emphasis on relationship with renal angiomyoadenomatous tumor. Am J Surg Pathol 2015;39:873–888.
Vang R, Levine DA, Soslow RA et al. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a rereview of cases lacking TP53 mutations in the Cancer Genome Atlas ovarian study. Int J Gynecol Pathol 2016;35:48–55.
The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–615.
Sircar K, Wang Z, Kim TB et al. Sarcomatoid clear cell RCC shows a distinct molecular pathogenesis and driver mutation profile. Mod Pathol 2016;29:262A–263A (abstract).
Calio A, Grignon DJ, Stohr BA et al. Renal cell carcinoma with TFE3 translocation and succinate dehydrogenase B mutation. Mod Pathol 2017;30:407–415.
Williamson SR, Kum JB, Goheen MP et al. Clear cell renal cell carcinoma with a syncytial-type multinucleated giant tumor cell component: implications for differential diagnosis. Hum Pathol 2014;45:735–744.
Klatte T, Rao PN, de Martino M et al. Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma. J Clin Oncol 2009;27:746–753.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Favazza, L., Chitale, D., Barod, R. et al. Renal cell tumors with clear cell histology and intact VHL and chromosome 3p: a histological review of tumors from the Cancer Genome Atlas database. Mod Pathol 30, 1603–1612 (2017). https://doi.org/10.1038/modpathol.2017.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.72
This article is cited by
-
New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)
-
Predominantly cystic clear cell renal cell carcinoma and multilocular cystic renal neoplasm of low malignant potential form a low-grade spectrum
Virchows Archiv (2018)
-
Reply to Chou et al ‘Do significant TFE3 gene rearrangements occur in succinate dehydrogenase deficient renal cell carcinoma? Borderline FISH results should be interpreted with caution’ Mod Pathol 2017; in press.
Modern Pathology (2017)
