
Abstract
We have previously shown that commonly expressed miRNAs influenced tumor molecular phenotype in colorectal cancer. We hypothesize that infrequently expressed miRNAs, when showing higher levels of expression, help to define tumor molecular phenotype. In this study, we examine 304 miRNAs expressed in at least 30 individuals, but in <50% of the population and with a mean level of expression above 1.0 relative florescent unit. We examine associations in 1893 individuals who have the tumor molecular phenotype data as well as miRNA expression levels for both carcinoma and normal colorectal tissue. We compare miRNAs uniquely associated with tumor molecular phenotype to the RNAseq data to identify genes associated with these miRNAs. This information is used to further identify unique pathways associated with tumor molecular phenotypes of TP53-mutated, KRAS-mutated, CpG island methylator phenotype and microsatellite instability tumors. Thirty-seven miRNAs were uniquely associated with TP53-mutated tumors; 30 of these miRNAs had higher level of expression in TP53-mutated tumors, while seven had lower levels of expression. Of the 34 miRNAs associated with CpG island methylator phenotype-high tumors, 16 were more likely to have a CpG island methylator phenotype-high tumor and 19 were less likely to be CpG island methylator phenotype-high. For microsatellite instability, 13 of the 22 infrequently expressed miRNAs were significantly less likely to be expressed in microsatellite unstable tumors. KRAS-mutated tumors were not associated with any miRNAs after adjustment for multiple comparisons. Of the dysregulated miRNAs, 17 were more likely to be TP53-mutated tumors while simultaneously being less likely to be CpG island methylator phenotype-high and/or microsatellite instability tumors. Genes regulated by these miRNAs were involved in numerous functions and pathways that influence cancer risk and progression. In summary, some infrequently expressed miRNAs, when expressed at higher levels, appear to have significant biological meaning in terms of tumor molecular phenotype and gene expression profiles.
Similar content being viewed by others


Main
Molecular pathological epidemiology is a growing field of study that utilizes molecular information from tumors to better understand disease processes and progression.1 Assessment of tumor molecular phenotype in colorectal cancer has led to a better understanding of lifestyle factors that are uniquely associated with specific tumor phenotype.2, 3, 4, 5, 6, 7, 8, 9, 10 Tumor markers also have been examined with survival in an effort to identify biomarkers that can be used to predict prognosis and provide individualized treatment.11, 12, 13, 14, 15, 16 While most studies have focused on common tumor molecular phenotype, such as TP53-mutated and KRAS-mutated tumors, microsatellite instability, and CpG Island Methylator Phenotype, studies are now examining other characteristics of tumors such as gene expression and miRNA expression that may be important in identifying key disease pathways.11, 14, 17, 18
MiRNAs are small, non-protein-coding RNA molecules that regulate gene expression either by post-transcriptionally suppressing mRNA translation or by causing mRNA degradation.18, 19, 20, 21, 22, 23 We have previously shown that commonly expressed miRNAs influence tumor molecular phenotype in colorectal cancer, with the greatest number of differentially expressed miRNAs being observed for microsatellite unstable tumors compared to microsatellite stable tumors.24 MiRNAs were less frequently differentially expressed for TP53-mutated tumors, KRAS-mutated tumors, and CpG island methylator phenotype-high tumors. Most research focusing on miRNAs and tumor phenotype have focused on microsatellite unstable and CpG island methylator phenotype-high tumors25 and on targeted miRNAs. Most targeted miRNAs studied, such as miR-21, are commonly expressed in tumors. Examination of infrequently expressed miRNAs may provide insight into unique pathways associated with tumor molecular phenotype.
In this study, we focus on miRNAs that are infrequently expressed in normal colorectal mucosa and carcinoma tissue. We have previously shown that 34.5% of miRNAs expressed in colon tumor tissue are expressed in fewer than 10% of the population.26 Almost half of the miRNAs expressed in colorectal cancer tissue are expressed in less than half of the population. This presents two interesting questions: first, are low levels of expression purely noise in the data representing background expression levels; second, are infrequently expressed miRNA meaningful when expressed at higher levels beyond what could be considered background noise? Since tumor molecular phenotype also varies in percentage of the population with a given phenotype, it is a logical question to determine if infrequently expressed miRNAs when expressed at higher levels are associated with unique tumor molecular phenotypes. In this study, we examine associations between tumor molecular phenotype and infrequently expressed miRNA to determine if such associations exist. We further examine infrequently expressed miRNAs to determine genes they may be associated with gene expression when expressed at higher levels along with functions and pathways associated with those genes. The size and design of this study makes in uniquely powered to examine the role of infrequently expressed miRNAs as they relate to colorectal cancer.
Methods
Study Participants
Study participants were recruited as part of two population-based case-control studies that included all incident colon and rectal cancer between 30 and 79 years of age who resided in Utah or were from the Kaiser Permanente Medical Care Program in Northern California. Participants were white, Hispanic, or black for the colon cancer study and also included participants of Asian race for the rectal portion of the study.27, 28 Case diagnosis was verified by the tumor registry data as a first primary adenocarcinoma of the colon or rectum and were diagnosed between October 1991 and September 1994 for the colon cancer study and between May 1997 and May 2001 for the rectal cancer study. Detailed study methods have been described.26 The study was approved by the Institutional Review Boards at the University of Utah and Kaiser Permanente Medical Care Program in Northern California.
RNA Processing
Formalin-fixed paraffin-embedded tissue from the initial biopsy or surgery was used to extract RNA. Both carcinoma tissue and adjacent normal mucosa were used. Tissue was micro-dissected from 1–4 sequential sections on aniline blue stained slides using an H&E slide for reference. Total RNA was extracted, isolated, and purified using the RecoverAll Total Nucleic Acid isolation kit (Ambion); NanoDrop spectrophotometer was used to determine RNA yields.
miRNA
The Agilent Human miRNA Microarray V19.0 containing probes for 2006 unique human miRNAs was used. The data were required to pass stringent quality control parameters established by Agilent to be included in the analyses. Quality control parameters included tests for excessive background fluorescence, excessive variation among probe sequence replicates on the array, and measures of the total gene signal on the array to assess low signal. If samples failed to meet quality standards for any of these parameters, the sample was re-labeled, hybridized to arrays, and re-scanned. If a sample failed quality control assessment a second time, the sample was deemed to be of poor quality and the sample was excluded from analysis. Our previous analysis has shown that the repeatability associated with this microarray was extremely high (r=0.98),26 and that comparison of miRNA expression levels obtained from the Agilent microarray to those obtained from qPCR had an agreement of 100% in terms of directionality of findings and that the fold change calculated for the miRNA expression difference between carcinoma and normal colonic mucosa was almost identical.29 Of the 2006 unique human miRNAs assessed on the Agilent microarray, 1226 were expressed in colon carcinoma tissue and 1179 in normal colon mucosa.
To normalize differences in miRNA expression that could be attributed to the array, amount of RNA, location on array, or factors that could erroneously influence miRNA expression levels, total gene signal was normalized by multiplying each sample by a scaling factor,30 which was the median of the 75th percentiles of all the samples divided by the individual 75th percentile of each sample.
mRNA: RNAseq Sequencing Library Preparation and Data Processing
Total RNA was run on 245 carcinoma and normal mucosa pairs; of these 217 paired samples passed quality control and were used in analyses. Tissues samples taken from the study subjects at time of diagnosis were used for RNA extraction as previously described.31 For mRNA analysis, RNA library construction was done with the Illumina TruSeq Stranded Total RNA Sample Preparation Kit with Ribo-Zero. The samples were then fragmented and primed for cDNA synthesis, adapters were then ligated onto the cDNA, and the resulting samples were then amplified using PCR; the amplified library was then purified using Agencount AMPure XP beads. A more detailed description of the methods can be found in our previous work.32 Illumina TruSeq v3 single read flow cell and a 50 cycle single-read sequence run was performed on an Illumina HiSeq instrument. Reads were aligned to a sequence database containing the human genome (build GRCh37/hg19, February 2009 from genome.ucsc.edu) and alignment was performed using novoalign v2.08.01. Total gene counts were calculated for each exon and UTR of the genes using a list of gene coordinates obtained from http://genome.ucsc.edu. We dropped features that were not expressed in our RNAseq data or for which the expression was missing for the majority of samples, retaining 17,384 protein-coding genes.32
Tumor Molecular Phenotype
We have previously assessed TP53 and KRAS mutations,4, 8, 33 the CpG island methylator phenotype using the classic panel that consisted of MLH1, CDKN2A and MINT1, MINT2 and MINT31,34 and microsatellite instability based on the mononucleotide repeats at BAT26 and TGFβR2 and a panel of 10 tetranucleotide repeats that were correlated highly with the Bethesda Panel;6 our original microsatellite instability studies were done prior to the development of the Bethesda Panel. Tumors were scored as CpG island methylator phenotype-high if two or more of the CpG islands were methylated for the five markers; otherwise they were classified as CpG island methylator phenotype-low/negative. This panel was run prior to the advent of more recent panels.35, 36
Statistical Methods
The study focuses on infrequently expressed miRNAs, which we define as being expressed in <50% of the study population for either normal mucosa or tumor. To be included in the analysis, miRNAs also had to have a mean level of expression of 1.0 Agilent Relative Florescent Unit (ARFU) in tumors or normal mucosa and be expressed in at least 30 individuals. Each infrequently expressed miRNA could be considered expressed or not in each tumor and normal, resulting in three primary dysregulation groups based on the tumor-normal expression differences: upregulated (expressed more in tumor than in normal), downregulated (expressed more in normal than in tumor), and referent (neither up- nor downregulated at the 25%tile/75%tile cutpoints). Rather than forcing the same number of subjects to fall into these three groups for all infrequently expressed miRNAs, cutpoints were selected based on the upper 25% and lower 25% of the tumor-normal differences for all infrequently expressed miRNAs. The resulting three-level dysregulation group factor (up, down, or referent) was used as a predictor in a per-miRNA logistic regression model also adjusting for age, study center, and sex and standardized what was considered true expression for all miRNAs. A total of 304 miRNAs were analyzed that fit these criteria. We used paired carcinoma and normal mucosa miRNA expression, evaluating differential expression between the two tissue types to control for differences in expression by tumor site and other potential confounding factors. Analyses were run separately for overall colorectal cancer, colon cancer, and rectal cancer. We analyzed difference in association for infrequently expressed miRNAs by TP53-mutated versus non-TP53-mutated, KRAS-mutated versus non-KRAS-mutated, CpG island methylator phenotype-high relative to CpG island methylator phenotype-low/negative, and microsatellite unstable compared to microsatellite stable. Adjustment for multiple comparisons was done using the positive false discovery rate Q value;37 given the infrequent expression of these miRNA, we report any associations for which the Q value was <0.05.
We compared those miRNA with a Q value of <0.05 (58=7 miRNAs) to the RNAseq data to identify genes whose expression was associated with these infrequently expressed miRNAs. To determine statistical significance between the miRNA::mRNA associations, we ran a Fisher–Pitman Monte Carlo test with 10 000 permutation comparing differences in mean levels of gene expression across miRNA dysregulation groups of ≤75 vs >75% in R using the ‘coin’ package. RPKM (Reads Per Kilobase of transcript per Million mapped reads) the mRNA expression level data were used in these analyses. Identification of networks and functions associated with genes whose mean expression was altered by miRNAs was done using Ingenuity Pathway Analysis; adjustments for multiple comparisons were made using the Benjamini and Hochberg method.38 Both causal and interaction networks were generated. Interaction networks were limited to 35 molecules per network and 25 networks per analysis, and excluded endogenous chemicals. We focused on algorithmically derived interaction networks, which are assigned a score based on their relevance to the genes in the input data set, the number of focus genes (that is, dysregulated genes in our data that are in that network), and their connectivity.39 The score is calculated as –log10P, where P is generated using a Fisher’s exact test.40 Studies have found scores >3 to be significant, with a score of 3 indicating a 1 in a 1000 chance that the focus genes are in a network due to random chance.41, 42, 43 Other studies have opted to utilize more stringent criteria and higher scores to ensure that their discovered networks are highly significant;44, 45 we utilized highly stringent criteria, only including networks with scores over 20.
Results
The study population is described in Table 1. Over half of the population were males. There were approximately equal numbers of individuals enrolled with proximal and distal colon tumors. Slightly less than half, 47.6%, of tumors had a TP53 mutation, 31.7% had a KRAS mutation, 21.2% were classified as CpG island methylator phenotype-high and 9.1% were microsatellite unstable.
Assessment of TP53-mutated tumors associated with infrequently expressed miRNAs showed that 30 miRNAs were more likely to have a TP53 mutation if they were upregulated in tumors, while seven miRNAs were associated with a lower likelihood of having a TP53 mutation if they were upregulated in tumors (Table 2). Most of the miRNAs (20 of the 37 miRNAs) were associated with a high level of differential expression in <20% of the. While some miRNAs were associated with a high level of differential expression in a large percentage of the population, these miRNAs were not expressed or extremely infrequently expressed in normal mucosa but were expressed to a greater degree in tumor tissue. There were no miRNAs more likely to have a TP53 mutation if downregulated after adjusting for multiple comparisons. Site-specific associations for colon and rectal cancer generally had Q values of >0.05. However, many of these miRNAs with a Q value of 0.03–0.04 overall had a Q value of 0.07 for colon cancer specifically, most likely reflecting the decrease in power when analyzing colon cancer specifically rather than colorectal cancer combined. The lowest Q values for miRNAs for rectal cancer were 0.083. There were no unique associations with KRAS-mutated tumors.
Thirty-five infrequently expressed miRNAs were associated with CpG island methylator phenotype-high tumors (Table 3). Of these, 35 miRNAs, 19 were less likely to be associated with a CpG island methylator phenotype-high tumor when upregulated in tumor tissue, while 16 were more likely to have a CpG island methylator phenotype-high tumor if the miRNA was upregulated in the tumor. Nine of these 35 miRNAs had over 20% of the population in the higher level of differential miRNA expression. As with TP53, many of these miRNAs had similar findings for colon cancer specifically as we observed for overall colorectal cancer, although the lowest FDR was 0.078 for colon cancer even when the raw P-values were <0.0001 and comparable for both overall colorectal cancer and colon cancer specifically. Also like for TP53, after adjustment for multiple comparisons there were no significant findings between CIMP-high tumors and downregulated miRNAs.
MSI was associated with 22 infrequently expressed miRNAs (Table 4). Of these miRNAs, the majority (13 of 22) were less likely to be associated with a microsatellite unstable tumor if upregulated in the tumor. Only two of the 22 miRNAs had over 20% of the population in the group of dysregulation. There were no significant associations with microsatellite unstable tumors and downregulated infrequently expressed miRNAs.
We determined which genes were associated with each of the 57 miRNAs that had a Q value of <0.05 using our RNAseq data. Those associations for all genes whose expression was altered by significant miRNAs are summarized in Supplementary Table 1. There was considerable overlap in miRNAs associated with tumor molecular phenotype. For instance, 19 miRNAs were associated with both CpG island methylator phenotype-high tumors and TP53-mutated tumors; 9 of these miRNAs also were associated with microsatellite unstable tumors. For each miRNAs where a higher level of expression increased the likelihood of having a TP53-mutated tumor, there was a decreased the likelihood of having a CpG island methylator phenotype-high or microsatellite unstable tumor.
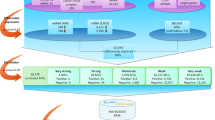
We have summarized the top three networks (Supplementary Table 2 has all networks with Scores of over 20) derived from genes linked to the 19 miRNAS that were associated with multiple tumor molecular phenotypes of TP53, CpG island methylator phenotype-high, and/or microsatellite unstable (Figure 1). Network 1 (immunological disease, inflammatory disease, and inflammatory response) had a Score of 28 and 35 focus molecules, including genes that were influenced by the miRNAs; Network 2 (cell cycle, cancer, cell-to-cell signaling and Interaction) had a Score of 25 and 34 Focus molecules influenced by the genes associated with these miRNAs; Network 3 (amino acid metabolism, small molecule biochemistry, drug metabolism) also had a Score of 25 and 34 focus molecules associated with genes linked to these miRNAs. The majority of genes in these networks were upregulated (indicated in red) when the miRNAs were expressed at higher levels. The genes that were downregulated (indicated in green NR3C1, TRPM6, GLP2R, ZFYVE28, FGD4, RNF112, TNFRSF17, TNFSF13, and CLEC3B) were all downregulated in the presence of high levels of miR-224-5p. Higher levels of miR-224-5p were more likely to be present in TP53-mutated tumors and less likely to be present in CpG island methylator phenotype-high tumors. PHGDH was upregulated at high levels of miR-19a-3p and KCND3 was upregulated at high levels of miR-424-5p; high levels of miR-424-5p were more likely to have a TP53-mutated tumor and less likely to have a CpG island methylator phenotype-high tumor. MYC expression was associated with six miRNA, miR-151a-3p, miR-19a-3p, miR-3687, miR-374b-5p, miR-4533, and miR-7-5p. Higher levels of miRNA expression for all but miR-4533 were associated with TP53-mutated tumors, while miR-4533 was associated with tumors that were more likely to have microsatellite instability and CpG island methylator phenotype-high.

Top Ingenuity Pathway Analysis networks associated with genes whose expression is altered by high levels of miRNA expression associated with both TP53 and CpG island methylator phenotypehigh and/or microsatellite instability.
Discussion
Our data suggest that some miRNAs, although infrequently expressed, when expressed at higher levels or upregulated, are associated with specific tumor molecular phenotype. We did not have similar associations for downregulated miRNAs. Of those infrequently expressed miRNAs, significantly associated with tumor molecular phenotype when expressed at high levels were more likely to be highly expressed in TP53-mutated tumors and less likely to be associated with CpG island methylator phenotype-high or microsatellite unstable tumors. Many of these miRNAs were associated with altered gene mRNA expression in colorectal cancer tissue when expressed at high levels.
Many miRNAs are expressed infrequently in the population and often have low levels of expression when detected.26 Many of the miRNAs that have levels of expression around 0 could be considered background noise from slight differences in RNA samples despite high quality control. Additionally, although the data were normalized, picking a scale to normalize on is arbitrary and a different scale could have slightly altered what was considered background levels of expression. The Agilent Platform that we used to collect miRNA data in this study has been noted as being able to detect low levels of expression.46, 47 On the basis of our findings, it appears that very low levels of expression are similar to no expression for most miRNAs, and that distinct associations for specific tumor molecular phenotype can only be seen when examining expression of these miRNAs at higher levels. These higher levels of expression are less likely to be the result of background expression, especially considering associations with tumor molecular phenotype.
To gain insight into pathways and functions of infrequently expressed miRNAs, we utilized our colorectal gene expression data from RNAseq. We assessed which genes were associated with miRNAs when miRNAs were more highly expressed. Since most of these miRNAs are infrequently expressed, there is less information regarding gene associations the existing databases, and even less information for colorectal tissue-specific expression, thus making use of our data imperative. Examining gene expression provided some insight into how these infrequently expressed miRNAs could be associated with various disease pathways. A limitation of RNAseq data, although a common method to determine miRNA::mRNA associations,48 is that miRNA targeted genes could be missed since gene expression studies more likely capture associations with transcription better than translation. However, we believe that our having RNAseq data in conjunction the miRNA data provides insight into colon-specific direct and indirect functions and pathways associated with these infrequently expressed miRNAs.
Given their infrequent expression, many of the miRNAs evaluated in our study have no known association with colorectal tumor molecular phenotype in the literature. However, our findings suggest that some infrequently expressed miRNAs, when they have high levels of expression in a tumor, may play an important role in tumorigenesis and the development of specific tumor phenotype. For instance, miR-19a-3p, which had about 25–30% of the population with high differential expression, was included previously in a miRNA cluster that functioned alongside Epstein–Barr virus to control gene expression in human B cells through a TP53-induced mechanism.49 While we could find no reported association between this miRNA, or the others evaluated in this study, and colorectal cancer-specific tumor molecular phenotype, these findings are consistent with our finding that high levels of miR-19a-3p is associated with a TP53 phenotype in colorectal cancer.
It has been shown that TP53 mutations are inversely related to CpG island methylator phenotype-high and microsatellite unstable in colorectal cancer; TP53 mutations are present in higher rates in microsatellite stable tumors, while CpG island methylator phenotype-high tumors also are frequently microsatellite unstable tumors.33, 50 Our findings support this pattern by demonstrating that certain infrequently expressed miRNAs when upregulated in TP53-mutated tumors are simultaneously more likely to be downregulated in CpG island methylator phenotype-high and microsatellite unstable tumors.
To further put these findings in perspective, we identified three major networks that represented the genes associated with those miRNAs that were upregulated in TP53-mutated tumors and downregulated in CpG island methylator phenotype-high and microsatellite unstable tumors. The first network has NR3C1 as one of its central components (See Figure 1). NR3C1 is a glucocorticoid receptor that induces apoptotic cell death, via decreased expression of anti-apoptotic proteins, such as BCL2 and MCL1, and induces expression of pro-apoptotic proteins like BCL2-like apoptosis initiator 11.51 In earlier studies, NR3C1 has been associated with proximal microsatellite unstable tumors, with hypermethylation of NR3C1 being identified as a marker for microsatellite unstable tumors and a marker to differentiate between CpG island methylator phenotype-high and CpG island methylator phenotype-low/negative phenotypes.52 These findings correlate with our identified association between the NR3C1 pathway and tumor phenotype; NR3C1 was downregulated in our data, suggesting less likely association with CpG island methylator phenotype-high and microsatellite unstable tumors. Our findings suggest that differential methylation of NR3C1, and its subsequent role in tumorigenesis and phenotype, may be in part due to the dysregulation of previously unstudied, infrequently expressed miRNAs.
The NFkB complex is central in our second Ingenuity Pathway Analysis network and is well known in literature for upregulating and promoting various proinflammatory cytokines and linking various gastrointestinal conditions such as inflammatory bowel disease, diabetes mellitus, and colorectal cancer.53 The classical NFkB pathway plays a major role in linking inflammation to the onset and progression of malignancy in various tissues.54 One pro,inflammatory stimulus includes red meat consumption, which has been linked to colon cancer and TP53-mutated tumors specifically.55, 56 A prospective study in Denmark has shown that the combination of polymorphisms in NFκB that downregulate its expression, and high red meat consumption increases the likelihood of developing colorectal cancer.57 They proposed that lower NFkB activity leads to higher loads of reactive oxygen species secondary to heme degradation, contributing to colorectal carcinogenesis. Moreover, other studies have found that the NFκB pathway to be linked with the TP53 pathway in hepatocellular carcinoma; the crosstalk between these two pathways is critical for the survival of HCC cells in the setting of reactive oxygen species.58 These previous findings further support an association between the NFkB complex and a TP53 molecular phenotype in certain cancers, especially in the setting of proinflammatory stimuli. Here we suggest that the upregulation of infrequently expressed miRNAs may provide an important link between NFκB and its related genes and TP53 phenotype in colorectal cancer.
In our third Ingenuity Pathway Analysis network, MYC encodes for c-myc, a transcription factor often constitutively amplified, leading to tumor progression of many cancers. In colorectal cancer, aberrant WNT/b-catenin pathway influences the amplification of MYC, leading to increased cellular proliferation.59 In our data, MYC was upregulated in conjunction with miRNAs that were upregulated in TP53-mutated tumors. Furthermore, the consensus molecular subtype 2 subtype of colorectal cancer is canonically known to have strong WNT/MYC activation in microsatellite stable tumors; this subtype was also found to be highly correlated with TP53-mutated tumors.60 This suggests that miRNA dysregulation from infrequently expressed miRNAs, may play an important role in MYC’s function in TP53-mutated molecular phenotype.
The study has several strengths and weaknesses. First, given the size of the study and the Agilent Platform used, we can identify and examine the impact of infrequently expressed miRNAs. Many studies are too small to be able to determine associations with infrequently expressed miRNAs. Our data set is rich, in that we have information on tumor molecular phenotype as well as RNAseq for a subset of these samples to improve our understanding of how miRNAs alter specific genes in colorectal tissue. One of the limitations of the study, which applies to the field of miRNA research, is the difficulty in understanding the pathways and genes associated with miRNA expression, especially when miRNAs alter multiple genes and genes are modified by multiple miRNAs. We have attempted to address this weakness in part by using our colorectal RNAseq data in conjunction with our miRNA data to identify genes that are up- or downregulated by infrequently expressed miRNAs. In this study, we have used adjacent tissue to the tumor as our comparison tissue. However, there are limitation that the 'normal' tissue is not true normal, although the best tissue available for comparison.
In summary, our data suggest that a large percentage of miRNAs expressed in colorectal tissue are infrequently expressed. However, some of the infrequently expressed miRNAs, when expressed at higher levels influence tumor molecular phenotype. This information is important for consideration pathways associated with cancer as well as examining lifestyle and environmental factors that may alter those pathways. Genes associated with these infrequently expressed miRNAs are involved in a variety of functions that may impact cancer development and prognosis.
Change history
21 September 2017
This article was originally published under NPG's License to Publish, but has now been made available under a [CC BY 4.0] license. The PDF and HTML versions of the paper have been modified accordingly.
References
Hamada T, Keum N, Nishihara R et al, Molecular pathological epidemiology: new developing frontiers of big data science to study etiologies and pathogenesis. J Gastroenterol 2016;52:265–275.
Nishihara R, Lochhead P, Kuchiba A et al, Aspirin use and risk of colorectal cancer according to BRAF mutation status. JAMA 2013;309:2563–2571.
Nishihara R, Wang M, Qian ZR et al, Alcohol, one-carbon nutrient intake, and risk of colorectal cancer according to tumor methylation level of IGF2 differentially methylated region. Am J Clin Nutr 2014;100:1479–1488.
Slattery ML, Anderson K, Curtin K et al, Lifestyle factors and Ki-ras mutations in colon cancer tumors. Mutat Res 2001;483:73–81.
Slattery ML, Anderson K, Curtin K et al, Dietary intake and microsatellite instability in colon tumors. Int J Cancer 2001;93:601–607.
Slattery ML, Curtin K, Anderson K et al, Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J Natl Cancer Inst 2000;92:1831–1836.
Slattery ML, Curtin K, Anderson K et al, Associations between dietary intake and Ki-ras mutations in colon tumors: a population-based study. Cancer Res 2000;60:6935–6941.
Slattery ML, Curtin K, Ma K et al, Diet activity, and lifestyle associations with p53 mutations in colon tumors. Cancer Epidemiol Biomarkers Prev 2002;541–548.
Slattery ML, Curtin K, Sweeney C et al, Diet and lifestyle factor associations with CpG island methylator phenotype and BRAF mutations in colon cancer. Int J Cancer 2007;120:656–663.
Bernstein AM, Song M, Zhang X et al, Processed and unprocessed red meat and risk of colorectal cancer: analysis by tumor location and modification by time. PLoS ONE 2015;10:e0135959.
Slattery ML, Herrick JS, Mullany LE et al, Improved survival among colon cancer patients with increased differentially expressed pathways. BMC Med 2015;13:75.
Samowitz WS, Curtin K, Ma KN et al, Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol Biomarkers Prev 2001;10:917–923.
Samowitz WS, Curtin K, Schaffer D et al, Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev 2000;9:1193–1197.
Dou R, Nishihara R, Cao Y et al, MicroRNA let-7, T cells, and patient survival in colorectal cancer. Cancer Immunol Res 2016;4:927–935.
Hanyuda A, Kim SA, Martinez-Fernandez A et al, Survival benefit of exercise differs by tumor IRS1 expression status in colorectal cancer. Ann Surg Oncol 2016;23:908–917.
Liao X, Morikawa T, Lochhead P et al, Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res 2012;18:2257–2268.
Ogino S, Meyerhardt JA, Kawasaki T et al, CpG island methylation, response to combination chemotherapy, and patient survival in advanced microsatellite stable colorectal carcinoma. Virchows Arch 2007;450:529–537.
Drusco A, Nuovo GJ, Zanesi N et al, MicroRNA profiles discriminate among colon cancer metastasis. PLoS ONE 2014;9:e96670.
Ambros V . The functions of animal microRNAs. Nature 2004;431:350–355.
Murray BS, Choe SE, Woods M et al, An in silico analysis of microRNAs: mining the miRNAome. Mol Biosyst 2010;6:1853–1862.
Arora S, Rana R, Chhabra A et al, miRNA-transcription factor interactions: a combinatorial regulation of gene expression. Mol Genet Genomics 2013;288:77–87.
Gartel AL, Kandel ES . miRNAs: Little known mediators of oncogenesis. Semin Cancer Biol 2008;18:103–110.
Nam S, Li M, Choi K et al, MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res 2009;37 (Web Server issue):W356–W362.
Slattery ML, Herrick JS, Mullany LE et al, Colorectal tumor molecular phenotype and miRNA: expression profiles and prognosis. Mod Pathol 2016;29:915–927.
Valeri N, Vannini I, Fanini F et al, Epigenetics, miRNAs, and human cancer: a new chapter in human gene regulation. Mamm Genome 2009;20:573–580.
Slattery ML, Herrick JS, Pellatt DF et al, MicroRNA profiles in colorectal carcinomas, adenomas and normal colonic mucosa: variations in miRNA expression and disease progression. Carcinogenesis 2016;37:245–261.
Slattery ML, Potter J, Caan B et al, Energy balance and colon cancer—beyond physical activity. Cancer Res 1997;57:75–80.
Slattery ML, Caan BJ, Benson J et al, Energy balance and rectal cancer: an evaluation of energy intake, energy expenditure, and body mass index. Nutr Cancer 2003;46:166–171.
Pellatt DF, Stevens JR, Wolff RK et al, Expression profiles of miRNA subsets distinguish human colorectal carcinoma and normal colonic mucosa. Clin Transl Gastroenterol 2016;7:e152.
Agilent Technologies I . Agilent GeneSpring User Manual Santa Clara. Aglient Technologies Inc: Santa Clara, CA, USA, 2013.
Slattery ML, Herrick JS, Mullany LE et al, An evaluation and replication of miRNAs with disease stage and colorectal cancer-specific mortality. Int J Cancer 2015;137:428–438.
Slattery ML, Pellatt DF, Mullany LE et al, Gene expression in colon cancer: A focus on tumor site and molecular phenotype. Genes Chromosomes Cancer 2015;54:527–541.
Samowitz WS, Holden JA, Curtin K et al, Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol 2001;158:1517–1524.
Samowitz WS, Albertsen H, Herrick J et al, Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology 2005;129:837–845.
Hinoue T, Weisenberger DJ, Lange CP et al, Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22:271–282.
Ogino S, Kawasaki T, Kirkner GJ et al, Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn 2007;9:305–314.
Storey JD . A direct approach to false discovery rates. J R Stat Soc B 2002;64 S:479–498.
Benjamini YH, Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 1995;57:289–300.
Savli H, Szendroi A, Romics I et al, Gene network and canonical pathway analysis in prostate cancer: a microarray study. Exp Mol Med 2008;40:176–185.
Li Y, Carrillo JA, Ding Y et al, Ruminal transcriptomic analysis of grass-fed and grain-fed angus beef cattle. PLoS ONE 2015;10:e0116437.
Baranzini SE, Galwey NW, Wang J et al, Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum Mol Genet 2009;18:2078–2090.
Yan-Fang T, Dong W, Li P et al, Analyzing the gene expression profile of pediatric acute myeloid leukemia with real-time PCR arrays. Cancer Cell Int 2012;12:40.
Naito Y, Kuroda M, Mizushima K et al, Transcriptome analysis for cytoprotective actions of rebamipide against indomethacin-induced gastric mucosal injury in rats. J Clin Biochem Nutr 2007;41:202–210.
Reyes-Gibby CC, Yuan C, Wang J et al, Gene network analysis shows immune-signaling and ERK1/2 as novel genetic markers for multiple addiction phenotypes: alcohol, smoking and opioid addiction. BMC Syst Biol 2015;9:25.
Jia P, Kao CF, Kuo PH et al, A comprehensive network and pathway analysis of candidate genes in major depressive disorder. BMC Syst Biol 2011;5 (Suppl 3):S12.
Sah S, McCall MN, Eveleigh D et al, Performance evaluation of commercial miRNA expression array platforms. BMC Res Notes 2010;3:80.
Mestdagh P, Hartmann N, Baeriswyl L et al, Evaluation of quantitative miRNA expression platforms in the microRNA quality control (miRQC) study. Nat Methods 2014;11:809–815.
Chou CH, Chang NW, Shrestha S et al, miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 2016 44:D239–D247.
Riley KJ, Rabinowitz GS, Yario TA et al, EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J 2012;31:2207–2221.
Bond CE, Umapathy A, Ramsnes I et al, p53 mutation is common in microsatellite stable, BRAF mutant colorectal cancers. Int J Cancer 2012;130:1567–1576.
Wei TT, Lin YT, Chen WS et al, Dual Targeting of 3-hydroxy-3-methylglutaryl coenzyme A reductase and histone deacetylase as a therapy for colorectal cancer. EBioMedicine 2016;10:124–136.
Ahlquist T, Lind GE, Costa VL et al, Gene methylation profiles of normal mucosa, and benign and malignant colorectal tumors identify early onset markers. Mol Cancer 2008;7:94.
Jurjus A, Eid A, Al Kattar S et al, Inflammatory bowel disease, colorectal cancer and type 2 diabetes mellitus: The links. BBA Clin 2016;5:16–24.
Li S, Pinard M, Wang Y et al, Crosstalk between the TNF and IGF pathways enhances NF-kappaB activation and signaling in cancer cells. Growth Horm IGF Res 2015;25:253–261.
Pellatt AJ, Slattery ML, Mullany LE et al, Dietary intake alters gene expression in colon tissue: possible underlying mechanism for the influence of diet on disease. Pharmacogenet Genomics 2016;26:294–306.
Slattery ML, Curtin K, Ma K et al, Diet activity, and lifestyle associations with p53 mutations in colon tumors. Cancer Epidemiol Biomarkers Prev 2002;11:541–548.
Andersen V, Christensen J, Overvad K et al, Polymorphisms in NFkB, PXR, LXR and risk of colorectal cancer in a prospective study of Danes. BMC Cancer 2010;10:484.
Huang Q, Zhan L, Cao H et al, Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016;12:999–1014.
Rennoll S, Yochum G . Regulation of MYC gene expression by aberrant Wnt/beta-catenin signaling in colorectal cancer. World J Biol Chem 2015;6:290–300.
Rodriguez-Salas N, Dominguez G, Barderas R et al, Clinical relevance of colorectal cancer molecular subtypes. Crit Rev Oncol Hematol 2017;109:9–19.
Acknowledgements
We acknowledge Sandra Edwards for data oversight and study management, and Michael Hoffman and Erica Wolff for miRNA analysis. We acknowledge Dr Bette Caan and the staff at the Kaiser Permanente Medical Research Program for sample and data collection. This study was supported by NCI grants CA163683 and CA48998.
Author contributions
MS obtained funding, planned study, oversaw study data collection and analysis, and wrote the manuscript; AP and FL helped interpret findings and helped write the manuscript; JS provided input into the statistical analysis; LM conducted bioinformatics analysis and helped write manuscript; RW oversaw laboratory analysis; WS reviewed manuscript and did pathology overview for the study; JH conducted statistical analysis and managed data. All authors approved final manuscript.
Disclaimer
The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Slattery, M., Lee, F., Pellatt, A. et al. Infrequently expressed miRNAs in colorectal cancer tissue and tumor molecular phenotype. Mod Pathol 30, 1152–1169 (2017). https://doi.org/10.1038/modpathol.2017.38
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.38
This article is cited by
-
The colorectal cancer epidemic: challenges and opportunities for primary, secondary and tertiary prevention
British Journal of Cancer (2018)
-
Utility of inverse probability weighting in molecular pathological epidemiology
European Journal of Epidemiology (2018)
-
Integration of pharmacology, molecular pathology, and population data science to support precision gastrointestinal oncology
npj Precision Oncology (2017)
