
Abstract
A subset of renal cell carcinomas shows TFEB overexpression secondary to MALAT1-TFEB gene fusion. As alternate mechanisms of TFEB overexpression are likely to have the same effect, we sought to determine the frequency of amplification of TFEB and the adjacent VEGFA gene at 6p21.1. As patients with metastatic renal cell carcinomas are managed with anti-VEGF therapies, we retrospectively assessed therapeutic response in patients with amplified tumors. Amplification status was analyzed for 875 renal cell carcinomas from our institution, a consultative case and 794 cases from The Cancer Genome Atlas. Cases were classified as having low level (5–10 copies), and high-level amplification (>10 copies), and were further analyzed for adjacent oncogene copy number status (n=6; 3 single-nucleotide polymorphism genomic microarray, 3 The Cancer Genome Atlas) and structural rearrangements (n=1; mate-pair sequencing). These were then reviewed for histopathology, immunophenotype, and response to VEGF-targeted therapy on follow-up. In all, 10/875 (1.1%) institutional cases, 1 consultative case, and 3/794 (0.4%) of The Cancer Genome Atlas cases showed TFEB high-level amplification, while 14/875 (1.6%) cases showed TFEB low-level amplification. All cases had associated VEGFA amplification. This was confirmed with evaluation for copy number changes (n=6). The 6p21.1 high and low-level amplified tumors occurred in adults (mean age: 66), with over half being ≥pT3 (13/25, 52%), and most showed oncocytic, tubulopapillary features and high grade (≥grade 3: 20/22, 91%). These were aggressive tumors with metastasis and death from renal cell carcinoma in 11 (of 24, 46%) cases. Four patients received targeted therapy and had a mean survival of 31 months (range: 17–50) post nephrectomy. In summary, a group of aggressive renal cell carcinomas show genomic amplification of the 6p21.1 region including TFEB and VEGFA genes and share morphologic features. Additional studies are warranted to determine whether these patients respond to anti-VEGF therapy.
Similar content being viewed by others

Main
Historically, renal cell carcinoma has been categorized into a few general subtypes. While specific genetic abnormalities have been known to occur with high frequency in these subtypes, the categorization had been largely based on morphologic features. Over the past several years, studies have shown that renal cell carcinoma consists of a much larger variety of subtypes with different genetic drivers, with many subtypes exhibiting distinct (albeit at times overlapping) histologic features, clinical course, and response to therapy.1 Tumors traditionally classified as papillary renal cell carcinoma in particular represent a heterogenous group of renal cell carcinomas requiring further definition.1 Indeed, within this group several distinct tumors associated with specific disease defining genetic abnormalities have been recently characterized, such as TFE3 translocation-associated renal cell carcinoma and hereditary leiomyomatosis and renal cell carcinoma-syndrome-associated renal cell carcinoma, and more are yet to be defined.1
The recently characterized TFEB-rearrangement-associated renal cell carcinoma is defined by a specific recurrent chromosomal translocation, t(6;11)(p21;q12) and is associated with distinct histologic and clinical features. The t(6;11) results in fusion of the untranslated metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) gene to the MITF transcription factor family member, transcription factor EB (TFEB) gene.2 This fusion leads to overexpression of TFEB.2 Conceptually, if overexpression of TFEB, at least in part, drives oncogenesis, alternate molecular mechanisms of increased TFEB expression, such as gene amplification, are likely to exist as well. Indeed, a recent study reported a single case of TFEB amplification, that also had a concurrent t(6;11)(p21;q12).3 This was followed by additional reports of TFEB amplification-associated renal cell carcinomas, including recent series of eight and three cases.4, 5, 6, 7 As the frequency of such amplification events is not known, we screened a large, subtype-specific, consecutive series of renal cell carcinomas from a single institution for TFEB amplifications by fluorescence in situ hybridization (FISH).
Gene amplification is well established as a significant mechanism of carcinogenesis and cancer progression, and it is often associated with high-level expression of the related protein products. The genes involved are typically associated with cell proliferation and cell cycle regulation such that overexpression results in uncontrolled tumor growth. While such amplification events confer a significant survival advantage to tumor cells, the tumor typically becomes addicted to these products. Therefore, gene amplifications offer a point of potential susceptibility for tumors and thus opportunity for therapy with agents targeting the protein products of the amplified gene or target proteins upon which they act.
Significantly, the TFEB gene lies immediately adjacent to other genes at 6p21.1 that are of known importance in oncogenesis such as RUNX2, CCND3, and VEGFA. Amplifications of vascular endothelial growth factor A (VEGFA), for instance, have been reported in a small but remarkably aggressive subgroup of colorectal cancers, while in osteosarcomas it is considered a predictor of poor tumor-free survival.8 Most importantly, advanced VEGFA-amplified hepatocellular carcinomas were found to be distinctly sensitive to sorafenib, a first-line drug that targets multiple kinases, including VEGF receptors (VEGFRs).9 In renal cell carcinomas, up to 30% of patients have metastases at presentation and management of metastatic disease relies on anti-angiogenic therapies with drugs such as sunitinib and pazopanib, as well as a number of other agents.10
The goal of our current study was to define the frequency of TFEB gene region amplification in renal cell carcinoma and to further characterize clinical and pathologic features. Given the proximity of TFEB and VEGFA, and the potential clinical implications of VEGFA amplifications, we also sought to determine the frequency of VEGFA gene amplification by FISH and to better define the region of amplification at chromosome 6p21.1 with the aid of single-nucleotide polymorphism genomic microarray and by mate-pair next-generation sequencing in a subset of amplified cases. Herein, we report the results of these findings and suggest that these results may have significant implications in treatment of some patients with aggressive renal cell carcinoma.
Materials and methods
Patient Specimens
The study consisted of subtype-specific, consecutive series of renal cell carcinomas from 875 patients diagnosed and treated by radical nephrectomy at a single institution between 1970 and 2012, including 394 patients with papillary renal cell carcinoma, 310 patients with clear cell renal cell carcinoma, 130 cases with chromophobe renal cell carcinoma, and 41 cases of renal cell carcinoma, unclassified as defined by the WHO classification. Apart from having carried a diagnosis of renal cell carcinoma and tissue available for analysis, cases were not otherwise selected for the study based on histopathologic or clinical criteria, and cases were a consecutive surgical series of renal cell carcinoma subtypes. Tissue microarrays were constructed from these cases using four, 1.0 mm cores of representative formalin-fixed, paraffin-embedded tissue from each tumor, and these tissue microarrays were utilized for screening FISH studies. Validation of FISH results as well as immunohistochemistry was performed on representative whole-slide sections. An additional case of papillary renal cell carcinoma, received in consultation from an external institution (PR, Department of Pathology, MD Anderson Cancer Center, Houston, Texas) as a part of routine clinical practice, was shown to have TFEB amplification, and was also included (876 total cases). Each case was independently reviewed for histologic features by three urologic pathologists (SG, JCC, and WRS).
Fluorescence In Situ Hybridization
Screening FISH studies were performed on unstained tissue slides from each tissue microarray using a laboratory developed, break-apart strategy probe for detection of TFEB rearrangement and a laboratory-developed VEGFA enumeration probe according to established methods.11, 12 Probe amplification was defined as the presence of an average of five or more copies of the probe per tumor nucleus. Cases were classified into two groups based on TFEB probe status: low-level amplification (5–10 copies) and high-level amplification (>10 copies). All cases identified as amplified by tissue microarray screening were subsequently shown to have TFEB and/or VEGFA amplification on reassessment of whole-tissue sections.
Immunohistochemistry
Immunohistochemistry for HMB45 (Dako, Carpinteria, CA, clone HMB45, 1:50), MelanA (Dako, clone A103, 1:50), Cytokeratin7 (CK7; Dako, clone OV-TL 12/30, 1:50-1:100), β-Catenin (Ventana Medical Systems, Tuscon, AZ, clone 14, prediluted), and E-Cadherin (Invitrogen, Carlsbad, CA, clone 4A2C7, prediluted) was performed on representative whole-tissue sections. β-Catenin and E-Cadherin expression was assessed to evaluate for Wnt signaling activation secondary to a DEK-ARMC12 rearrangement.
Single-Nucleotide Polymorphism Genomic Microarray
To better define the nature of the amplicons identified by FISH studies, two cases shown to have high-level amplification of both TFEB and VEGFA and one case showing TFEB high-level amplification only were also analyzed by single-nucleotide polymorphism genomic microarray (Oncoscan, Affymetrix). Briefly, formalin-fixed, paraffin-embedded tumor tissue was macrodissected from unstained slides and the genomic DNA then extracted. After DNA quantitation by Qubit, samples were processed for the Affymetrix OncoScan platform following the manufacturer’s recommended protocol. Data files were analyzed utilizing the Affymetrix ChAS software.
Mate-Pair Sequencing
A single case shown to have TFEB/VEGFA amplification by FISH and microarray analysis was subjected to mate-pair next-generation genetic sequencing. In brief, DNA was extracted from snap-frozen fresh tissue stored at −80 °C and mate-pair next-generation genetic sequencing libraries were assembled from whole-genome DNA using the Illumina Nextera MP library preparation kit and were sequenced on the Illumina HiSeq 2500 in rapid run mode as previously described.13 Pooled libraries were sequenced using 101-basepair reads and paired end sequencing. Paired reads were mapped to the human GRCh38 reference genome using 32-bit binary indexing of the genome as previously described.13 Discordant mate-pair reads mapping >15 kb apart or in different chromosomes were selected for further analysis. A mask was used to eliminate common variants and discordant fragments from experimental or algorithmic errors.
Literature Review and Data Extraction From the Cancer Genome Atlas Project
The publicly available cBioPortal.32e34 platform was used to analyze data from The Cancer Genome Atlas project related to renal cell carcinomas for copy number alterations of the TFEB and VEGFA genes and led to the identification of three TFEB-VEGFA-co-amplified renal cell carcinomas (two papillary renal cell carcinoma and one clear cell renal cell carcinoma) from a cohort of 794 cases (clear cell renal cell carcinoma: 448; papillary renal cell carcinoma: 280; chromophobe renal cell carcinoma: 66). Thirteen other cases were previously reported in the literature, with known amplifications of TFEB and without documentation of VEGFA amplification status.3, 4, 5, 6, 7 A single case showed concurrent t(6;11)(p21;q12) rearrangement, without VEGFA amplification status having been reported.3
Statistical Analysis
Frequency counts and percentages were used to analyze continuous clinicopathologic variables. Statistical significance, where applicable, was assessed with tests that were two-sided, with P<0.05 considered to be statistically significant.
Results
Renal Cell Carcinoma With High-Level 6p21.1 Amplification
We identified a total of 10 (of 875; 1.1%) cases of renal cell carcinoma with high-level (>10 copies) TFEB amplification (papillary renal cell carcinoma: 7/394, 1.8%; clear cell renal cell carcinoma: 1/310, 0.3%; chromophobe renal cell carcinoma: 0/130, 0%; renal cell carcinoma, unclassified: 3/41, 7.3%) through screening of institutional tumor tissue microarrays by FISH for TFEB (Table 1, cases 1–9 and 11). We also identified an additional case from an outside institution (Table 1, case 10). FISH for VEGFA copy number showed co-amplification at a level equivalent to that of TFEB in nine (of 10) cases. One case showed high-level amplification of TFEB without high-level amplification of VEGFA, although FISH did show an overall gain in VEGFA copy number of approximately five copies per nucleus on average (low-level amplification).
The clinical and pathologic features for our cases of renal cell carcinoma with high-level 6p21.1 amplification are summarized in Tables 1 and 2. The average age at diagnosis was 67 years (range 34–83) with a slight male predominance (eight males vs three females). Most tumors were originally classified as papillary renal cell carcinoma by histologic assessment (7/11; 64%). Associated diseases and neoplasia were documented to screen for potential syndromic associations; however, no consistently associated conditions were identified.
The tumor size ranged from 1.8 to 18.5 cm (mean 8.5 cm) and the majority were tumor stage pT3 or higher (7/11; 64%). By histopathologic examination, most tumors showed a distinct appearance with predominantly tubulopapillary architecture (Figure 1; Table 2, 10/11, 91%). Areas with a more nested growth were also common as either a primary or secondary growth pattern, and tumor cells were primarily oncocytic (10/11; 91%), with clear cell morphology as a prominent finding in some cases (Supplementary Figure 1). Characteristic histopathologic features, which have been reported for t(6;11)(p21;q12)-associated renal cell carcinomas, include a biphasic morphology. Similar, distinct biphasic morphology was not a prominent feature of high-level amplified tumors, with only three cases showing areas reminiscent of biphasic morphology (Supplementary Figure 2). No definitive TFEB rearrangement was detected in these cases, in addition to amplification, as was described in a single case by Peckova et al.3 The tumors were characterized by adverse histologic features with almost all showing a high International Society of Urologic Pathology grade and half showing tumor necrosis. However, no cases showed sarcomatoid features and only a single case showed rhabdoid features.

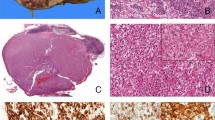
Histopathology: tubulopapillary pattern (Cases 7, 8, 9, and 10). Representative H&E-stained images of TFEB-VEGFA (6p21.1)-co-amplified renal cell carcinomas showing oncocytic, tubulopapillary architecture ((a) Case 8; (b), Case 9; × 100 magnification). Tubulopapillary features in a background of nested ((c) Case 10; × 200 magnification) as well as both oncocytic and clear cell areas are depicted ((d) Case 7; × 100 magnification). TFEB, transcription factor EB; VEGFA, vascular endothelial growth factor A.
Immunohistochemistry for CK7 was performed in all 11 cases and for HMB45 and MelanA in 10 cases (Table 3). CK7 immunostaining was identified in greater than half of cases, commonly in a focal to patchy pattern, and was associated with areas with tubulopapillary architecture (6 of 11, 55%). MelanA expression was identified in a majority of cases (7 of 10, 70%; Supplementary Figure 3); however, HMB45 expression was less common and identified in only a single case (1 of 10, 10%). Overall, these findings are similar to three cases identified in The Cancer Genome Atlas data set and are consistent with what has been reported in the literature3, 4, 5, 6, 7 (Supplementary Tables 1b).
Renal Cell Carcinoma With Low-Level 6p21.1 Amplification
Prior studies have proposed a cutoff of >10 copies of TFEB.6 However, as 6p21.1-amplified renal cell carcinomas are a recently described entity, the cutoff for amplification, as identified by FISH, still needs to be defined.3, 4, 5, 6, 7 Therefore, we screened tissue microarrays for low-level TFEB amplification (5–10 copies), in order to characterize them and compare these cases to a cohort of TFEB high-level amplification cases (>10 copies), with all cases being managed uniformly at a single institution. A total of 14 (of 875, 1.6%) cases of renal cell carcinoma with TFEB low-level amplification were identified (papillary renal cell carcinoma: 8/394, 2%; clear cell renal cell carcinoma: 2/310, 0.7%; chromophobe renal cell carcinoma: 3/130, 2.3%; renal cell carcinoma, unclassified: 1/41, 2.4%; Supplementary Table 1a). In all cases, VEGFA copy number was roughly equivalent to that of TFEB by FISH.
With regard to clinical and pathologic features, a similar trend was noted in cases with low-level 6p21.1 amplification as that seen in those with high-level amplification. The average age at diagnosis was 66 years (range 46–81), and 6 of 14 cases (43%) were staged as at least pT3a, with an average tumor size of 9 cm. Sarcomatoid and rhabdoid changes were rare, and these tumors were high International Society of Urological Pathology grade and half exhibited necrosis (Supplementary Tables 1a and 2a). Similarly, patchy CK7 expression (6 of 14, 43%) and rare HMB45 staining (1 of 14, 7%) was seen. In contrast to cases with high-level amplification, rare positivity for MelanA (2 of 14, 14%; Supplementary Figure 4) was identified (Supplementary Table 3a).
Renal Cell Carcinoma With 6p21.1 Amplification (All Cases Combined, ≥5 Copies)
As cases with TFEB high-level amplification (n=11) and low-level amplification (n=14) exhibited similar clinicopathologic features, the key characteristics of all combined cases (n=25) have been summarized here (Tables 1,2,3). Tumors were predominantly diagnosed as papillary renal cell carcinoma (60%; high-level amplification: 63.6%, low-level amplification: 57.1%, P=0.75). The average age at diagnosis was 66 years (high-level amplification: 67, low-level amplification: 66, P=0.75), and 52% of cases were staged as at least pT3a (high-level amplification: 64%, low-level amplification: 43%, P=0.32), with an average tumor size of 8.8 cm (high-level amplification: 8.5 cm, low-level amplification: 9.0 cm, P=0.78). Rhabdoid change was seen in 12% (high-level amplification: 9.1%, low-level amplification: 14.3%, P=0.70) and sarcomatoid change in 4% (high-level amplification: 0%, low-level amplification: 7.1%, P=0.38). These tumors were high International Society of Urological Pathology grade (90.9%; high-level amplification: 81.8%, low-level amplification: 100%, P=0.15) and half exhibited necrosis (48%; high-level amplification: 45.5%, low-level amplification: 50%, P=0.83). Immunophenotypic characterization revealed a similar pattern of expression for CK7 (48%; high-level amplification: 54.5%, low-level amplification: 42.9%, P=0.58) and HMB45 (8.3%; high-level amplification: 10%, low-level amplification: 7.1%, P=0.81). However, MelanA expression did reveal a statistically significant difference (37.5%; high-level amplification: 70%, low-level amplification: 14.3%, P=0.01, Fisher’s exact test).
Amplicon Assessment by Single-Nucleotide Polymorphism-Based Genomic Microarray
Microarray analysis was performed in two cases shown to have high-level TFEB/VEGFA co-amplification and the one case with only high-level TFEB amplification associated with low-level amplification of VEGFA, and showed a structurally complex amplicon in all cases (Figure 2 and Figure 3a). All cases showed amplification of a region extending from approximately position 41 283 633 to 48 027 731 (6.7 megabases) on the short arm of chromosome 6 (Figure 3a). In addition, all cases showed amplification of discontinuous regions adjacent to this area extending further in both centromeric and telomeric directions. No classic cytogenetic abnormalities associated with papillary, clear cell, or chromophobe renal cell carcinoma were identified in these cases.

Histopathology and FISH (Case 1). Representative H&E-stained images of a TFEB-VEGFA (6p21.1)-co-amplified renal cell carcinoma showing high-grade papillary architecture with both oncocytic and clear cell morphology ((a) ×100 magnifications; (b) ×400 magnifications). Representative images of FISH for VEGFA (c) and TFEB (d) have been depicted. FISH, fluorescence in situ hybridization; TFEB, transcription factor EB; VEGFA, vascular endothelial growth factor A.

Single-nucleotide polymorphism genomic microarray and mate-pair sequencing (Cases 7, 8, and 9). A schematic representation of the area of amplification on chromosome 6p21.1 by single-nucleotide polymorphism genomic microarray (a) and of the structural rearrangement between DEK and ARMC12 genes (b) have been depicted. In a, the solid red line depicts the locus of TFEB amplification in Cases 7, 8, and 9. Cases 7 and 8 exhibited TFEB/VEGFA high-level amplification, compared to Case 9, which showed high-level amplification of TFEB and low-level amplification for VEGFA. The locus of VEGFA amplification has been indicated by a dotted line. TFEB, transcription factor EB; VEGFA, vascular endothelial growth factor A.
Mate-Pair Sequencing
Two adjacent areas of amplification were identified on chromosome 6p in three consecutive cases analyzed by single-nucleotide polymorphism genomic microarray- Case 7 and 8 (unlike Case 9, which demonstrated an isolated TFEB (6p21.1) high-level amplification with only low-level amplification of VEGFA (6p21.1)). As this pattern of amplification (Figure 3a) was suggestive of an underlying structural gene rearrangement, mate-pair sequencing was pursued, with a specific focus on the short arm of chromosome 6. This was done for Case 8, which had documented high-level co-amplifications of TFEB-VEGFA by both FISH and single-nucleotide polymorphism microarray. This revealed prominent amplification of the TFAP2B gene, a known transcriptional regulator of VEGF signaling (Supplementary Figure 5). On the basis of prior studies, high level evidence supporting structural rearrangements was defined as junctions greater than 30 kb with at least three unique supporting fragments.13 A structural gene rearrangement, with a total of 59 unique mapped fragments, between the DEK gene on 6p22.3 and the ARMC12 gene on 6p21.31 spanning a length of 17.5 megabases was identified (Figure 3b), and this was confirmed by sanger sequencing (data not shown). As DEK-ARMC12 rearrangements are implicated in Wnt signaling, the activation of this pathway was assessed by immunostaining for β-catenin and E-Cadherin (not shown). No nuclear localization of β-catenin was seen in all 15 cases that were tested (high-level amplification: five cases; low-level amplification: 10 cases). Loss of membranous cell surface localization of E-Cadherin was seen for 13 of 15 cases (high-level amplification: three of five cases; low-level amplification: 10 of 10 cases).
Clinical Follow-Up
The aggressive nature of these tumors is supported by regional lymph node involvement and distant metastasis at the time of radical nephrectomy in many of these patients (Table 4 and Supplementary Table 4a). Overall, 11 (of 24, 46%) patients developed regional and distant metastasis, over a mean follow-up duration of 101 months, with no significant difference between patients with high-level amplification and low-level amplification (high-level amplification: 5/10, 50%; low-level amplification: 6/14, 43%, P=0.74). Common sites of distant involvement included lung, bone, vagina, and soft tissue, and all patients with advanced disease died of disease-related complications. This includes Case 20; this patient died from surgical complications at the time of nephrectomy. Five-year cancer-specific survival rates post nephrectomy were 44% (four of nine cases) and 50% (seven of fourteen cases), respectively, for TFEB-VEGFA-co-amplified renal cell carcinomas with high- and low-level amplifications (P=0.80; Figure 4a). Cancer-specific survival for all cases combined at 5 years of follow-up post nephrectomy was 48% (11 of 23 cases, Figure 4b).

Cancer-specific survival rates. Cancer-specific survival rates (95% confidence interval, number still at risk) at 5 years following surgery are 44% ((a) four of nine cases) for TFEB-VEGFA-co-amplified renal cell carcinoma (high-level amplification), 50% ((a) 7 of 14 cases) for TFEB-VEGFA-co-amplified renal cell carcinoma with low-level amplification. Five-year survival rates for all cases combined are 48% ((b) 11 of 23 cases). TFEB, transcription factor EB; VEGFA, vascular endothelial growth factor A.
Importantly, four patients received VEGF-targeted therapy, which included three with high-level and one with low-level TFEB-VEGFA co-amplifications. Of the patients with the high-level co-amplifications, the first (Case 8) was initially diagnosed with pT3c Nx disease (vena caval involvement) and Sutent (sunitinib) was initiated at 38 months of follow-up post nephrectomy, after documentation of brain, lung, adrenal, and lymph node metastasis. However, this was discontinued after 20 days because of decompensation secondary to gastrointestinal bleeding. Targeted therapy was not re-initiated and the patient died shortly thereafter. Owing to the advanced stage of disease, and limited duration of therapy, it is difficult to draw any definitive conclusions regarding therapeutic efficacy from this case study.
A second patient (Case 7) diagnosed with pT2a N1 disease (para aortic lymph node involvement) received Votrient (pazopanib) after documentation of bone metastasis (inferior pubic ramus and left 7th rib), at ∼5 months of follow-up post nephrectomy. After initiation of therapy, the patient survived for 7.5 months, following which he died from progressive renal cell carcinoma including malignant pleural effusion and ascites. Similarly, a third patient (Case 11) with pT3c N0 disease went on to develop vertebral body, retroperitoneal, and right lower quadrant abdominal wall metastases, ∼8 months following radical nephrectomy. Sutent (sunitinib) was initiated ∼1 month thereafter, with the patient surviving for an additional 8 months before dying of renal cell carcinoma.
Finally, a fourth patient (Case 25) diagnosed with pT3b, N1, M1 disease at nephrectomy due to the presence of interaortocaval lymph node involvement and liver metastases had low-level TFEB-VEGFA co-amplifications. He was managed with Sutent (sunitinib) starting ∼19 months post nephrectomy. This patient survived for ∼31 months thereafter, during which time he was managed with temsirolimus and Nexavar (sorafenib), as well. This patient appears to have had an excellent response to his therapy. According to the current (2017) American Cancer Society Statistics, 5-year mortality rates for patients with metastatic renal cancer are as low as 12% (ref. 14). Given the aggressive nature of advanced disease, identification of more such cases is needed to assess therapeutic benefit.
Discussion
Using a combination of a FISH-based strategy supplemented with single-nucleotide polymorphism-based genomic microarray and mate-pair sequencing, we identified 25 renal cell carcinomas with amplification of TFEB/VEGFA region after screening a total of 876 renal cell carcinoma cases. As these cases were not otherwise preselected based on histopathologic or clinical features, our estimate for the incidence of TFEB-VEGFA-amplified renal cell carcinomas is ∼3.8% for papillary renal cell carcinoma (15/394), 2.3% for chromophobe renal cell carcinoma (3/130), 1.0% for clear cell renal cell carcinoma (3/310), and 9.8% for our institutional cohort of unclassified renal cell carcinomas (4/41). In addition, bioinformatics analysis of publicly available The Cancer Genome Atlas data sets revealed an incidence of 0.7% for papillary renal cell carcinoma (2/280), 0.2% for clear cell renal cell carcinoma (1/448), and 0% for chromophobe renal cell carcinoma (0/66). Taken together, the highest combined incidence is in cases initially classified as papillary renal cell carcinoma (2.5%, 17 of 674), followed by chromophobe renal cell carcinoma (1.5%, 196) and clear cell renal cell carcinoma (0.5%, 4 of 758). Cases with TFEB/VEGFA high-level amplification alone had the highest combined incidence in papillary renal cell carcinomas as well (9 of 674, 1.3%).
Our findings in the current study appear concordant with those in recent series by Argani et al and Williamson et al6, 7 with regards to morphology (including architecture and grade) and clinical course. On re-review, most cases (19 of 25, 76%) were documented to exhibit morphologic features characterized by prominent tubulopapillary architecture, oncocytic cytoplasm, and high International Society of Urological Pathology grade. While adverse histopathologic features such as rhabdoid and sarcomatoid change were infrequent, a large percentage of these cases demonstrated high International Society of Urological Pathology grade (≥grade 3: 20 of 22 cases, 91%) and coagulative tumor necrosis (12 of 25, 48%), suggesting poor prognosis. Only three (of 25, 12%) cases showed some histologic overlap (histology reminiscent of biphasic features) with the classic morphology of t(6;11)(p21;q12)-associated renal cell carcinomas.6 In addition, no classic cytogenetic abnormalities associated with papillary renal cell carcinoma, clear cell renal cell carcinoma, or chromophobe renal cell carcinoma were identified in three cases analyzed by single-nucleotide polymorphism genomic microarray, further supporting the hypothesis that these cases likely represent a unique entity.
The average size of the tumors was 8.8 cm (range, 1.8–22), with 19 (of 25, 76%) patients presenting with pT2 disease or higher and 13 (of 25, 52%) patients presenting with pT3 disease or higher. The aggressive nature of these tumors was borne out by the documentation of regional and distant metastasis in 11 (of 24, 46%) patients over a mean follow-up duration of 101 months, with all patients with aggressive disease dying of renal cell carcinoma. Five-year cancer-specific survival was 44% (9 cases) and 50% (14 cases) for TFEB-VEGFA-co-amplified renal cell carcinomas with high and low-level amplifications, and 48% (23 cases) for all cases combined. This is significantly lower than what has been reported for chromophobe renal cell carcinoma (89%, 151 cases), papillary renal cell carcinoma (91%, 448 cases), and clear cell renal cell carcinoma (74%, 2101 cases) in our experience.15 In addition, this is in contrast to the behavior of t(6;11) renal cell carcinomas, which are relatively more indolent and metastasize less frequently, with metastatic disease reported in approximately five (of 60, 8%) cases, in one study.6
In our current study, results of single-nucleotide polymorphism-based genomic microarray in three cases and mate-pair sequencing in one case suggest that the amplicon involved in this TFBEB/VEGFA amplification event may be defined by a minimal ‘critical’ region of ∼8 megabases within a region of the p12.1 and p21.1 bands on chromosome 6. However, all tumors also showed additional areas of genomic amplification and copy number gain in 6p12.1 to 6p21.1 region adjacent to the primary region of amplification. This finding is significant because many genes within this region have been implicated in oncogenesis, with the primary mechanism being overexpression of their protein product. Oncogenes contained within this region include FOXP4, TFEB, CCND3, ABCC10, PTK7, VEGFA, HSP90AB1, RUNX2, CLIC5, and MEP1A. Interestingly, a similar minimal region of gain on the short arm of chromosome 6, mapping to 6p22 as well as structural rearrangements with a common translocation breakpoint at 6p22, has been implicated in the development of retinoblastomas.16 Other than the well-defined association between TFEB and renal cell carcinomas, prior studies have documented an association between aneuploidy, high TNM stage, high grade, high proliferation, young age, and Cyclin D3 expression in renal cell carcinomas as well.17, 18 The phenomenon of 6p21 amplification, with RUNX2 amplifications, has been documented in osteosarcomas and is associated with poor response to chemotherapy.19, 20, 21, 22 High expression of RUNX2 has also been implicated in metastatic prostate and breast cancers.19, 23, 24 FOXP4 has been implicated to have a role in non-small cell lung cancer and has been found to be involved in translocations associated with cancer.25, 26, 27 HSP90AB1 is the only other potentially targetable gene (using drugs such as geldanamycin).28 Amplification of the TFAP2B gene, a member of the AP2 transcription factor family, was identified in Case 8 on mate-pair sequencing. This is particularly noteworthy, as recent studies have shown that TFAP2B has a significant role in the development of non-small cell lung cancer through mechanisms that involve VEGF-dependent signaling.29
Defining when a genomic gain constitutes amplification of a particular gene of interest is a complex issue. There is no universal definition and thus defining amplification is often done on a tumor-by-tumor basis. Ultimately, the threshold at which a genomic gain becomes significant needs to be defined based on clinical information, such as response to targeted therapy and requires significant supporting data. Without a basis in this tumor type upon which to define amplification, almost any threshold used to define amplification would be arbitrary and defining amplification in the current study such that some cases with biologically significant copy number gains were artificially excluded. Therefore, we chose to define tumors into those with low-level amplification, with 5–10 copies of TFEB and/or VEGFA, and those with high-level amplification, with >10 copies, based on the definition set forth by Argani et al.6 The results of our study suggest that there are tumors with 5–10 copies of TFEB/VEGFA with similar characteristics to tumors with >10 copies. It is worth noting that we did find a significantly higher frequency of MelanA expression in the tumors with high level vs low-level amplification. It may be that high-level amplification contributes more to activation of the TFEB-associated pathway relative to low-level amplification. When considering that some low-level amplified tumors also showed MelanA expression, this difference may indicate that our low-level amplification group contains some tumors in which the amplification of the region is significant to the pathogenesis and others in which it is less or not significant to pathogenesis. Further studies are required to resolve this question and we anticipate that in the future the definition of 6p21.1-amplified tumors is likely to evolve, before it is standardized.
At present, targeted therapy with tyrosine kinase inhibitors that target the VEGFR is approved for patients with metastatic renal cell carcinoma, including patients undergoing cytoreductive nephrectomy. Although our study was too small to draw conclusions regarding the effectiveness of VEGFR-targeted therapies in these patients, the one patient with prolonged response to treatment as well as the experience of VEGFR-targeted therapy in other tumor types with VEGFA amplification suggest that future studies are needed to examine the effectiveness of VEGFR-targeted therapies in patients with 6p21.1-amplified renal cell carcinomas. Morphologic features such as prominent tubulopapillary architecture, oncocytic cytoplasm, and high grade could aid in the selection of cases for molecular testing. Although it must be noted, there is morphological heterogeneity in these tumors such that capturing all amplified tumors will require testing of all cases.
In our current study, mate-pair sequencing also revealed concurrent structural rearrangements on the short arm of chromosome 6 leading to a fusion of the DEK and ARMC12 genes in one case. DEK gene fusions (with NUP214) were first described in acute myeloid leukemia.30 Subsequently, overexpression of DEK was identified in bladder cancer, breast cancer, glioblastomas, hepatocellular carcinoma, melanoma, retinoblastoma, colorectal cancer, as well as cancers of the oral cavity and gynecologic tract.16, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42 Functionally, overexpression of DEK promotes cellular proliferation by promoting replication fork progression and neoplastic transformation both in vitro and in vivo, and this is partially attributed to the protection of daughter cells from DNA damage.43, 44, 45 The specific mechanism of action involves preferential binding to transcription start sites of highly transcribed genes in areas of hypomethylation.46 In addition, recent studies have shown that DEK signaling promotes both the production and secretion of Wnt ligands to promote canonical β-catenin signaling.36, 47 The DEK fusion partner ARMC12 is an Armadillo (Arm) repeat-containing protein, which is the homolog of mammalian β-catenin, and these have been implicated in canonical β-catenin signaling through the regulation of β-catenin stability and interaction with adherens junctions, as well.48 No evidence of increased Wnt signaling was identified based on a lack of nuclear β-catenin localization. Loss of cell surface localization of E-Cadherin was seen for 13 of 15 (87%) cases; however, the significance of this finding is uncertain as both papillary and clear cell renal cell carcinoma have low reported rates of E-Cadherin expression.49 As a DEK-ARMC12 fusion has not been previously reported, and was identified in just a single case, both its prevalence and functional significance in contributing to renal cell carcinoma oncogenesis is uncertain. It is entirely possible that it represents a nonspecific structural abnormality secondary to amplification-related events in the region.7, 50 However, future studies will be needed to address the significance of this finding.
In summary, our current study builds on the recently reported work of Argani et al and Williamson et al,6, 7 with a total of 25 new cases of TFEB-amplified renal cell carcinomas. Our results suggest that 6p21.1-amplified renal cell carcinomas are a distinct entity, and that these tumors present at higher stage, are higher grade and have a worse clinical outcome than typical papillary, clear cell, and chromophobe renal cell carcinoma.6 Our findings indicate that these tumors are defined by amplification of a region of chromosome 6p21.1 including not only TFEB but also VEGFA and a number of other oncogenes. Further studies are warranted to determine whether these tumors may be susceptible to targeted therapy.
References
Linehan WM, Spellman PT, Ricketts CJ et al, Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 2016;374:135–145.
Davis IJ, Hsi BL, Arroyo JD et al, Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci USA 2003;100:6051–6056.
Peckova K, Vanecek T, Martinek P et al, Aggressive and nonaggressive translocation t(6;11) renal cell carcinoma: comparative study of 6 cases and review of the literature. Ann Diagn Pathol 2014;18:351–357.
Durinck S, Stawiski EW, Pavia-Jimenez A et al, Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 2015;47:13–21.
Williamson SR, Grignon DJ, Favazza L et al, Chromosome 6p amplification including the TFEB gene: a novel mechanism of renal cell carcinoma pathogenesis? Mod Pathol 2016;29;270A–271A.
Argani P, Reuter VE, Zhang L et al, TFEB-amplified renal cell carcinomas: an aggressive molecular subset demonstrating variable melanocytic marker expression and morphologic heterogeneity. Am J Surg Pathol 2016;40;1484–1495.
Williamson SR, Grignon DJ, Cheng L et al, Renal cell carcinoma with chromosome 6p amplification including the TFEB gene: a novel mechanism of tumor pathogenesis? Am J Surg Pathol 2016;41;287–298.
Yang J, Yang D, Sun Y et al, Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer 2011;117;4925–4938.
Horwitz E, Stein I, Andreozzi M et al, Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov 2014;4;730–743.
Motzer RJ, Hutson TE, Cella D et al, Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369;722–731.
Hodge JC, Pearce KE, Wang X et al, Molecular cytogenetic analysis for TFE3 rearrangement in Xp11.2 renal cell carcinoma and alveolar soft part sarcoma: validation and clinical experience with 75 cases. Mod Pathol 2014;27;113–127.
Sukov WR, Hodge JC, Lohse CM et al, TFE3 rearrangements in adult renal cell carcinoma: clinical and pathologic features with outcome in a large series of consecutively treated patients. Am J Surg Pathol 2012;36;663–670.
Drucker TM, Johnson SH, Murphy SJ et al, BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics 2014;30;1627–1629.
American Cancer Society. Cancer facts & figures 2017. American Cancer Society. Available at www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf; accessed on 01/27/2017.
Lohse CM, Gupta S, Cheville JC . Outcome prediction for patients with renal cell carcinoma. Semin Diagn Pathol 2015;32;172–183.
Paderova J, Orlic-Milacic M, Yoshimoto M et al, Novel 6p rearrangements and recurrent translocation breakpoints in retinoblastoma cell lines identified by spectral karyotyping and mBAND analyses. Cancer Genet Cytogenet 2007;179;102–111.
Hedberg Y, Roos G, Ljungberg B et al, Cyclin D3 protein content in human renal cell carcinoma in relation to cyclin D1 and clinico-pathological parameters. Acta Oncol 2002;41;175–181.
Pertia A, Nikoleishvili D, Trsintsadze O et al, Immunoreactivity of p27(Kip1), cyclin D3, and Ki67 in conventional renal cell carcinoma. Int Urol Nephrol 2009;41;243–249.
Ito Y, Bae SC, Chuang LS . The RUNX family: developmental regulators in cancer. Nat Rev Cancer 2015;15;81–95.
Lau CC, Harris CP, Lu XY et al, Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes Cancer 2004;39;11–21.
Martin JW, Zielenska M, Stein GS et al, The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma 2011;2011;282745.
Sadikovic B, Thorner P, Chilton-Macneill S et al, Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer 2010;10;202.
Akech J, Wixted JJ, Bedard K et al, Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 2010;29;811–821.
Pratap J, Wixted JJ, Gaur T et al, Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res 2008;68;7795–7802.
Howarth KD, Blood KA, Ng BL et al, Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene 2008;27;3345–3359.
Lam EW, Brosens JJ, Gomes AR et al, Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer 2013;13;482–495.
Yang T, Li H, Thakur A et al, FOXP4 modulates tumor growth and independently associates with miR-138 in non-small cell lung cancer cells. Tumour Biol 2015;36;8185–8191.
Yamaki H, Nakajima M, Shimotohno KW et al, Molecular basis for the actions of Hsp90 inhibitors and cancer therapy. J Antibiot 2011;64;635–644.
Fu L, Shi K, Wang J et al, TFAP2B overexpression contributes to tumor growth and a poor prognosis of human lung adenocarcinoma through modulation of ERK and VEGF/PEDF signaling. Mol Cancer 2014;13;89.
von Lindern M, Breems D, van Baal S et al, Characterization of the translocation breakpoint sequences of two DEK-CAN fusion genes present in t(6;9) acute myeloid leukemia and a SET-CAN fusion gene found in a case of acute undifferentiated leukemia. Genes Chromosomes Cancer 1992;5;227–234.
Datta A, Adelson ME, Mogilevkin Y et al, Oncoprotein DEK as a tissue and urinary biomarker for bladder cancer. BMC Cancer 2011;11;234.
Grasemann C, Gratias S, Stephan H et al, Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene 2005;24;6441–6449.
Khodadoust MS, Verhaegen M, Kappes F et al, Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res 2009;69;6405–6413.
Kondoh N, Wakatsuki T, Ryo A et al, Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res 1999;59;4990–4996.
Kroes RA, Jastrow A, McLone MG et al, The identification of novel therapeutic targets for the treatment of malignant brain tumors. Cancer Lett 2000;156;191–198.
Privette Vinnedge LM, McClaine R, Wagh PK et al, The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene 2011;30;2741–2752.
Carro MS, Spiga FM, Quarto M et al, DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle 2006;5;1202–1207.
Han S, Xuan Y, Liu S et al, Clinicopathological significance of DEK overexpression in serous ovarian tumors. Pathol Int 2009;59;443–447.
Lin L, Piao J, Gao W et al, DEK over expression as an independent biomarker for poor prognosis in colorectal cancer. BMC Cancer 2013;13;366.
Martinez-Useros J, Rodriguez-Remirez M, Borrero-Palacios A et al, DEK is a potential marker for aggressive phenotype and irinotecan-based therapy response in metastatic colorectal cancer. BMC Cancer 2014;14;965.
Nagpal JK, Das BR . Identification of differentially expressed genes in tobacco chewing-mediated oral cancer by differential display-polymerase chain reaction. Eur J Clin Invest 2007;37;658–664.
Wu Q, Li Z, Lin H et al, DEK overexpression in uterine cervical cancers. Pathol Int 2008;58;378–382.
Sanden C, Nilsson HJ, Gullberg U . The DEK oncoprotein is upregulated by multiple leukemia-associated fusion genes. Blood Cells Mol Dis 2015;54;284–285.
Wise-Draper TM, Mintz-Cole RA, Morris TA et al, Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res 2009;69;1792–1799.
Deutzmann A, Ganz M, Schonenberger F et al, The human oncoprotein and chromatin architectural factor DEK counteracts DNA replication stress. Oncogene 2015;34;4270–4277.
Sanden C, Jarvstrat L, Lennartsson A et al, The DEK oncoprotein binds to highly and ubiquitously expressed genes with a dual role in their transcriptional regulation. Mol Cancer 2014;13;215.
Privette Vinnedge LM, Benight NM, Wagh PK et al, The DEK oncogene promotes cellular proliferation through paracrine Wnt signaling in Ron receptor-positive breast cancers. Oncogene 2015;34;2325–2336.
Coates JC . Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol 2003;13;463–471.
Langner C, Ratschek M, Rehak P et al, Expression of MUC1 (EMA) and E-cadherin in renal cell carcinoma: a systematic immunohistochemical analysis of 188 cases. Mod Pathol 2004;17;180–188.
Kalyana-Sundaram S, Shankar S, Deroo S et al, Gene fusions associated with recurrent amplicons represent a class of passenger aberrations in breast cancer. Neoplasia 2012;14;702–708.
Acknowledgements
We would like to thank Tammy L Torgerson and the Mayo Clinic Cytogenetics Core for providing assistance with FISH, and Christine M Lohse and Janis L Donovan for administrative assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Gupta, S., Johnson, S., Vasmatzis, G. et al. TFEB-VEGFA (6p21.1) co-amplified renal cell carcinoma: a distinct entity with potential implications for clinical management. Mod Pathol 30, 998–1012 (2017). https://doi.org/10.1038/modpathol.2017.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.24
This article is cited by
-
Unveiling the genetic and epigenetic landscape of colorectal cancer: new insights into pathogenic pathways
Medical Oncology (2023)
-
Past, present, and future perspectives of transcription factor EB (TFEB): mechanisms of regulation and association with disease
Cell Death & Differentiation (2022)
-
TFE3 and TFEB-rearranged renal cell carcinomas: an immunohistochemical panel to differentiate from common renal cell neoplasms
Virchows Archiv (2022)
-
Diagnostic utility of one-stop fusion gene panel to detect TFE3/TFEB gene rearrangement and amplification in renal cell carcinomas
Modern Pathology (2021)
-
New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}