
Abstract
Myelodysplastic syndromes (MDS) are a heterogeneous group of diseases with varying genetic aberrations. Half of MDS patients have normal karyotype, obscuring the underlying condition indicating a need for new markers for improved diagnostics and prognosis. We performed a retrospective review of sequential MDS patients who underwent chromosomal genetic array testing (CGAT) between November 2008 and March 2014. Total Genomic Aberration (TGA) scores, with and without copy-neutral loss of heterozygosity (cnLOH), were compared to pathology and clinical data. Of 68 MDS participants, 50 patients (73%) had abnormal CGAT results. 32% showedcnLOH, 41% had no cnLOH but displayed copy number aberration (CNAs). Of 26 patients with normal cytogenetics, 46% had clonal abnormalities by CGAT. Abnormal CGAT results were associated with lower overall survival (P=0.04). Overall survival in patients with TGA above the median (68.6 Mb) was significantly inferior to those below the median (HR=2.9, 95% CI=1.3-6.8, P=0.01). Furthermore, there was an observed association between increased TGA and increased dysplastic lineages (Ptrend=0.003). CGAT studies provide important findings that extend beyond current standard testing. Clinical utility of CGAT includes improved diagnostic yield, correlation of extent of TGA and increased dysplastic features, and survival.
Similar content being viewed by others

Main
Myelodysplastic syndromes are a group of clonal hematopoietic stem cell disorders characterized by cytopenias, morphologic dysplasia, and an increased risk of acute myeloid leukemia.1 The incidence of myelodysplastic syndrome is, on average, 3–5/100 000 with increasing rates in older patients. Current standards and guidelines for myelodysplastic syndrome are primarily those referenced in the 2008 Edition of the WHO Classification of Haematopoietic and Lymphoid Tissue Textbook with updates published in 2016.2, 3, 4 Despite recent advances in diagnostic modalities, myelodysplastic syndrome continues to show variability in its clinical course for prognosis and response to treatment, indicating the need for further subclassification.5, 6, 7 Difficulties in establishing dysplastic features especially in the early stages, accurate counting of blast percentages, as well as the lack of uniformity in cytogenetic and molecular analysis across institution are some of the reasons for variability.7
Currently, the only curative treatment for myelodysplastic syndrome is allogeneic hematopoietic cell transplantation.8 The use of the International Prognostic Scoring System, now in a revised version, has aided physicians in identifying patients who may or may not benefit from hematopoietic cell transplantation.9 The new Revised International Prognostic Scoring System takes into consideration marrow blast percentage, peripheral blood counts and cytogenetic findings, of which the latter has the most profound impact on prognosis. However, 40–50% of myelodysplastic syndrome patients have a normal karyotype,10 therefore obscuring the prognostic value by the Revised International Prognostic Scoring System and indicating a need for new markers for improved diagnostics and prognosis.
Copy neutral loss of heterozygosity describes a phenomenon whereby one of two homologous chromosomal regions is lost, but various mechanisms have ensured the presence of two identical copies of such region in the genome. As a result, the karyotype appears normal or ‘copy neutral.’ Copy neutral loss of heterozygosity and microdeletions in myelodysplastic syndrome have been well described in the 2008 and 2016 WHO Editions of myelodysplastic syndrome classification.11, 12 Single-nucleotide polymorphism arrays are consistent and reliable in finding regions of copy neutral loss of heterozygosity and for review of the whole genome for copy numbers. Copy number aberrations can be detected more accurately by array testing than by routine karyotyping13 and fluorescence in situ hybridization (FISH). Total genomic aberration numbers, including both acquired copy neutral loss of heterozygosity and copy number aberrations, can be calculated from single-nucleotide polymorphism array data. Potential mechanisms of copy neutral loss of heterozygosity include mitotic recombination, gene conversion, or trisomy rescue in somatic cells11, 14, 15 serving as the second hit in the Knudson two-hit tumorigenesis hypothesis. Copy neutral loss of heterozygosity can lead to duplication of an activating mutation in an oncogene, deletion or loss of function of a tumor suppressor gene, and duplication or deletion of a methylation allele that regulates gene expression.15 The presence of copy neutral loss of heterozygosity in the genome of myelodysplastic syndrome cells appear to portend a worse prognosis for the patient.16, 17 However, additional studies to evaluate the significance of copy neutral loss of heterozygosity in relation to morphologic features and the clinical course are needed.
Our goal in this study was to evaluate the significance of copy number aberrations and copy neutral loss of heterozygosity in myelodysplastic syndrome by correlating our findings with clinical characteristics, immunophenotypes, morphologic abnormalities, and outcome data. We hypothesize that chromosome genomic array testing studies will show that total genomic aberration numbers positively correlate with dysplastic features/lineage involvement and impacts survival.
Materials and methods
Patients and Biologic Materials
Patients who were diagnosed with myelodysplastic syndrome (WHO 2008) and myelodysplastic syndrome/myeloproliferative neoplasms unclassifiable at the University of Washington Medical Center and/or Seattle Cancer Care Alliance, and underwent chromosome genomic array testing were included in this study. All patients signed informed consents and the Fred Hutchinson Cancer Research Center Institutional Review Board approved the study. Pertinent clinical information was reviewed, which included the following: presentation of disease; associated comorbidities; select laboratory data; transplant parameters; and relapse and survival.
Hematopathology Review, Flow Cytometry, Molecular Diagnostic Data
Pathology slides were reviewed by a hematopathologist and morphologic dysplastic features categorized according to those listed in the 2008 Edition of the WHO Classification of Haematopoietic and Lymphoid Tissue textbook.18 Clinical chart reviews were performed by a clinical oncologist with expertise in myelodysplastic syndrome and, when data were available, Revised International Prognostic Scoring System scores were calculated. Relapse and survival data were retrospectively captured in May 2016.
Ten-color multiparameter flow cytometry was performed on bone marrow aspirates obtained as routine baseline assessment; details have been described previously.19, 20 Data were collected from samples with copy neutral loss of heterozygosity, and specific blast immunophenotype and other pertinent findings were recorded.
Molecular Diagnostics
Molecular diagnostic testing with PCR-based single-gene assays were performed as part of routine clinical management in a subset of these patients. The following genes were included: JAK2; BCR/ABL1; FLT3; NPM1; and CEBPA.
Targeted gene panel next-generation sequencing data were available in the form of Oncoplex reports (University of Washington, Genomics and Molecular Pathology) as part of the diagnostic workup. Specific methodology has been previously published.21 Specific myelodysplastic syndrome-related genes included in this 194-gene panel were as follows: TP53; RUNX1; ETV6; TET2; DNMT3A; ASXL1; EZH2; IDH1/2; SF3B1; SRSF2; U2AF1; ZRSR2; NRAS; CBL; JAK2; and SETBP1 (for full gene list please see: http://tests.labmed.washington.edu/UW-OncoPlex).
Conventional Cytogenetics and FISH
Bone marrow aspirate samples from all patients were tested for cytogenetic abnormalities using standard culturing and G-banding technique at the Seattle Cancer Care Alliance. Karyotype designation was based on the International System for Human Cytogenetic Nomenclature.22 FISH was performed at the Seattle Cancer Care Alliance according to the standard procedures. FISH probes were purchased from Abbott Molecular (Abbott Park, IL) and Cytocell–Rainbow Scientific (Windsor, CT).
Chromosomal Genomic Array Testing
DNA was extracted from fresh bone marrow aspirates and from frozen marrow samples using Qiagen Puregene (Germantown, MD) according to the manufacturer’s protocol. Genomic DNA microarray CytoScan HD, with probes for both copy number and single-nucleotide polymorphisms, was purchased from Affymetrix (Santa Clara, CA). The criteria used to identify an aberration were a minimum of 100 kb and 25 probes for copy number aberrations and 10 Mb for terminal copy neutral loss of heterozygosity (13 Mb for interstitial copy neutral loss of heterozygosity). Total genomic aberrations were calculated based on total length of DNA in Mb of somatic aberration (copy neutral loss of heterozygosity and copy number aberrations).
Statistical Analyses
On the basis of chromosome genomic array testing results, our study population was separated into three groups for statistical comparison: (1) patients with copy neutral loss of heterozygosity (with or without additional chromosome genomic array testing abnormalities); (2) patients with abnormal chromosome genomic array testing but no copy neutral loss of heterozygosity; and (3) patients who had a normal chromosome genomic array testing study.
In addition to chromosome genomic array testing results, other characteristics that were examined included the following: morphology; immunophenotype; mutation results; FISH; conventional cytogenetic data; and outcome parameters such as relapse and survival. Comparisons among chromosome genomic array testing groups were performed using the Kruskal–Wallis test. Linear regression analysis was used to assess trends in total genomic aberrations as a function of number of dysplastic lineages. Overall survival was estimated using the Kaplan–Meier method. Relapse was estimated using cumulative incidence estimates, with non-relapse mortality as a competing risk. Cox regression analysis was used to assess univariate prognostic factors for relapse and survival. Follow-up time as of 25 May 2016 ranged from 790 to 2715 days.
Five patients had no follow-up after sample collection and are not included in the survival analyses. Five additional patients were known to have died without a precise date of death. Two of these were assigned death dates using the date of the clinic note stating that the patient was deceased; the other three were assigned death dates of 9 months after sample collection, which was the median time to death among patients with known dates.
Results
Population Characteristics
We tested bone marrow samples from 68 patients with myelodysplastic syndrome and indeterminate myelodysplastic syndrome/myeloproliferative neoplasms, evaluated from November 2008 through March 2014 by chromosome genomic array testing. Clinical characteristics are summarized in Table 1. Clinical data were incomplete in 12 of 68 patients. For 58 patients, we could calculate the Revised International Prognostic Scoring System scores, confirming 37 patients were high risk and 21 patients with low-risk disease. Transplant data included in Table 1 reflects the patient characteristic at the time of sample collection. During follow-up, 11 patients relapsed and 27 patients died.
Chromosome Genomic Array Testing Data Compared to Cytogenetics by Karyotype and FISH
Of 68 total patients, 38 (56%) had abnormal cytogenetics (by karyotype and FISH), 26 (38%) had normal cytogenetics (by karyotype and FISH), and in 4 patients (5.8%) karyotyping was unsuccessful. Table 2 summarizes the chromosome genomic array testing results in comparison to cytogenetic data. By chromosome genomic array testing, 50 of 68 patients showed an abnormal result (73%). Among these, 22 (32%) showed copy neutral loss of heterozygosity, whereas 28 (41%) showed no copy neutral loss of heterozygosity but did have abnormal copy numbers (copy number aberrations). In all, 18 patients (28%) had normal chromosome genomic array testing results.
Of the 26 samples with normal cytogenetics, 12 (46%) showed clonal abnormalities only detected by chromosome genomic array testing. Of the 38 samples with abnormal cytogenetics, chromosome genomic array testing provided additional information for copy neutral loss of heterozygosity abnormalities in 13 samples (33%) and small submicroscopic copy number aberrations in 10 samples (26%). In the 4 samples, which failed to grow in culture, 2 (50%) showed isolated copy neutral loss of heterozygosity.
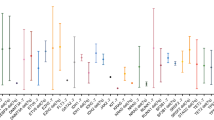
The most common regions of copy number aberrations were very similar to those described in myelodysplastic syndrome, including del 5q, monosomy 7, trisomy 8, and del 20q (Figure 1a). Gain of 1q was also prevalent, as seen in 5 patients. The most frequently noted copy neutral losses of heterozygosity, in descending order, were copy neutral loss of heterozygosity of 9p (n=8), 11q and 17p (n=3 each), 4q, 11p, and 17q (n=2 each). Single cases of 5q, 7q, and 14q copy neutral loss of heterozygosity were also observed. Furthermore, 8 samples with 9p copy neutral loss of heterozygosity showed myelofibrotic changes, and 5 showed unilineage dysplasia. Diagnoses included myelodysplastic syndrome with marrow fibrosis (n=4), chronic myelomonocytic leukemia with marrow fibrosis (n=1), and myelodysplastic syndrome secondary to polycythemia vera with cytogenetic clonal evolution (n=3). All samples with 17p copy neutral loss of heterozygosity occurred in the context of 5q deletions (in addition to other chromosomal aberrations) with multilineage dysplasia. Five 9p copy neutral loss of heterozygosity cases had JAK2 V617F mutations (Figure 1b), 2 were negative for JAK2 mutation by Oncoplex NGS, and 1 could not be tested.

(a) The most common regions of copy number aberration (copy number aberrations) and copy neutral loss of heterozygosity from all patients in this study. (b) Allelic tracks of representative patients with 9p copy neutral loss of heterozygosity. (c) Diagram of one potential mechanism of 9p copy neutral loss of heterozygosity resulting from mitotic recombination in somatic cells of myelodysplastic syndrome/myeloproliferative neoplasms patients. (a) A summary composite of the regions of chromosomal aberration in the 68 patients included in our cohort. In the top row are copy number aberrations, with blue representing areas of gains and red representing areas of losses, the bottom row is the single-nucleotide polymorphism track with golden areas representing areas of copy neutral loss of heterozygosity. (b) Allelic tracks of patients with 9p copy neutral loss of heterozygosity depicting various size of the copy neutral loss of heterozygosity and the percentage of cells abnormal. The top panel shows copy neutral loss of heterozygosity of the entire short arm of chromosome 9 in 100% of cells. The second panel shows 9p copy neutral loss of heterozygosity in ~20% of cells. The bottom panel shows copy neutral loss of heterozygosity of the terminal 9p in 70–80% of cells in a post-transplant patient. The location of the JAK2 gene is marked by the red dotted line. (c) Diagram of one potential mechanism of 9p copy neutral loss of heterozygosity resulting from mitotic recombination in somatic cells of myelodysplastic syndrome/myeloproliferative neoplasms patients with JAK2 mutation. The normal chromosome 9 homolog is shown in blue and the JAK2 mutated chromosome 9 homolog in red. The JAK2 mutation is depicted by the black bar. Daughter cells with 9p copy neutral loss of heterozygosity and homozygous JAK2 mutation would have the selective growth advantage in vivo. The difference in the crossover point of the mitotic recombination may lead to differences in the size of the copy neutral loss of heterozygosity seen in different patients, as shown in b.
Chromosome Genomic Array Testing and Blast Percentage (Morphology and Flow Cytometry)
Morphology of the erythroid, myeloid, and megakaryocyte lineages was evaluated, and dysplasia was called out when more than 10% of the lineage cells met the 2008 WHO criteria.18 Table 3 shows a significant correlation between total genomic aberrations of abnormal chromosome genomic array testing results and dysplastic morphology (Ptrend=0.05 for abnormal chromosome genomic array testing samples with copy neutral loss of heterozygosity, and Ptrend=0.003 for all abnormal chromosome genomic array testing samples). In a separate analysis (data not shown), we noted that patients with abnormal chromosome genomic array testing had higher blast percentages by flow cytometry, but this association did not reach statistical significance (P=0.07). No significant association existed between abnormal immunophenotypic markers, including CD4, CD5, CD7, CD13, CD15, CD33, CD34, CD38, CD45, CD56, CD117, CD123, and HLA-DR, and chromosome genomic array testing results.
Clinical follow-up information to perform a survival analysis was available in 63 patients. When dividing this group of patients into three categories—those with normal chromosome genomic array testing vs patients with abnormal chromosome genomic array testing with or without copy neutral loss of heterozygosity—the latter two categories had worse overall survival (P=0.04; Figure 2). The median overall survival for patients whose myelodysplastic syndrome features only copy neutral loss of heterozygosity was 24.9 months, when compared to patients whose myelodysplastic syndrome were abnormal by chromosome genomic array testing but did not show copy neutral loss of heterozygosity, the latter group of patients has a longer overall survival of 35.8 months. For patients whose chromosome genomic array testing was interpreted as ‘normal’ the median overall survival was not reached. We also assessed whether the extent of chromosome genomic array testing abnormalities mattered. Patients with total genomic aberrations above the median of 68.6 Mb (range, 0–592) had inferior overall survival compared to those below the median (mortality hazard ratio=2.9, 95% CI, 1.3–6.8, P=0.01; Figure 3).

Kaplan–Meier estimates show a statistically significant survival difference between patients with abnormal copy neutral loss of heterozygosity and patients with normal chromosome genomic array testing (overall P=0.04). The individual comparisons were P=0.04 with copy neutral loss of heterozygosity, and P=0.05 without copy neutral loss of heterozygosity (using normal as the reference).

Kaplan–Meier estimates of all patients showed a significant survival difference between patients with total genomic aberrations values above and below the median (mortality hazard ratio=2.9, 95% CI, 1.3–6.8, P=0.01).
Because Cluzeau et al.23 previously reported that total genomic aberrations >100 Mb were associated with worse survival among high-risk myelodysplastic syndrome patients, we specifically evaluated the high-risk patients in our cohort. Among 36 patients with Revised International Prognostic Scoring System >3 with survival data available, 14 had total genomic aberrations <100 and 22 had total genomic aberrations >100.
Patients with total genomic aberrations >100 Mb had worse overall survival than those with total genomic aberrations <100 Mb (mortality hazard ratio=3.0, 95% CI, 1.0–9.3, P=0.05; Figure 4). There was no evidence of an impact of total genomic aberrations among the low-risk patients, but the sample size was small (n=21) and, there were only three deaths in this category. The median total genomic aberrations in the low-risk patients was 0.7 Mb (range, 0–305.7).

Among patients with high-risk myelodysplastic syndrome based on Revised International Prognostic Scoring System, Kaplan–Meier estimates showed a survival advantage for those with total genomic aberrations <100 Mb (mortality hazard ratio=3.0, 95% CI, 1.0–9.3, P=0.05).
Thirty-seven of the 63 patients (59%) received transplants at a median of 117 days (range, 20–1030 days) after sample collection. The proportion of patients transplanted was similar among the groups defined above and adjustment for transplant as a time-dependent covariate had no material impact on the results.
There was no significant association of total genomic aberrations and relapse but sample size was small—only 11 patients relapsed. One patient had a normal chromosome genomic array testing study, 4 showed copy neutral loss of heterozygosity, and 6 showed an abnormal chromosome genomic array testing study without copy neutral loss of heterozygosity.
Among 9 patients with myelodysplastic syndrome, marrow samples were submitted for concurrent Oncoplex testing. Of these, no mutations were seen in 4 samples; 1 case had low tumor burden and next-generation sequencing was not performed; and mutations were present in the remaining 4 patients (Table 4). Mutations included the most commonly mutated genes associated with myelodysplastic syndrome, such as SF3B1, SRSF2, ASXL1, and TET2. In 1 case with 4q copy neutral loss of heterozygosity, there was a TET2 splicing variant occurring at 96% allelic frequency consistent with a homozygous abnormality due to copy neutral loss of heterozygosity.
Discussion
The first myelodysplastic syndrome case series utilizing array chromosomal genomic hybridization was published by Paulsson et al.24 Publications since then have established the improved diagnostic yield of this approach as compared to conventional cytogenetics.25, 26, 27, 28 New technology has also been added, including flow cytometry, chromosome genomic array, and next-generation sequencing. However, the clinical utility of these modalities, especially in regards to their correlation with classical morphology-based diagnosis of myelodysplastic syndrome and with patient outcome, remains to be characterized. Here we investigated sequential patients with myelodysplastic syndrome who underwent chromosome genomic array testing and we performed an in-depth retrospective analysis of chromosome genomic array testing correlation with pathologic and clinical characteristics to determine the impact of copy neutral loss of heterozygosity and total genomic aberrations on survival. Utilizing total genomic aberrations as a quantitative measure of cytogenomic abnormality, we compared morphology and immunophenotype to clinical outcomes and demonstrated important clinical utility of array testing in myelodysplastic syndrome.
The association between dysplastic features and increasing total genomic aberrations is a novel observation, which suggests that the more morphologic dysplasia a marrow sample displays the more extensive the underlying genomic perturbation reflected by higher total genomic aberrations. Both copy number aberrations and copy neutral loss of heterozygosity contributed significantly to this correlation in the current study, highlighting the utility of the single-nucleotide polymorphism-containing array platforms. When comparing morphologic findings with the quantitative measure of total genomic aberrations from chromosome genomic array testing (Table 3) a significant parallel trend was seen. Samples with fewer dysplastic lineage (0 or 1) showed lower total genomic aberrations while samples with more dysplastic lineages (2 or 3) tended to yield higher total genomic aberrations numbers (Ptrend 0.003). However, when considering samples with copy neutral loss of heterozygosity alone, a borderline trend was observed among the dysplastic lineages (Ptrend 0.05). It may be that this result reflects lower-grade myelodysplastic syndromes, such as that used by the 2008 WHO classification system, which classifies myelodysplastic syndrome into unilineage vs those with multilineage dysplasia. Although our initial hypothesis of a linear relationship between total genomic aberrations and dysplastic lineage was not confirmed, there was a trend for 0, 1, or 2 (not for 3) dysplastic lineages and increasing total genomic aberrations. Immunophenotype analysis by flow cytometry showed no significant differences when we compared the patients with a normal chromosome genomic array testing study to those with an abnormal chromosome genomic array testing, with and without copy neutral loss of heterozygosity.
Our data suggest that chromosome genomic array testing can be an effective risk stratification tool. When considering the three groups of patients—abnormal chromosome genomic array testing with copy neutral loss of heterozygosity, abnormal chromosome genomic array testing without copy neutral loss of heterozygosity, and normal chromosome genomic array testing—a statistically significant survival difference was seen (Figure 2). Furthermore, we have shown better survival in myelodysplastic syndrome patients with total genomic aberrations below the median (68.6 Mb) in our cohort (Figure 3). Of the high-risk subset based on Revised International Prognostic Scoring System scores, we demonstrated that patients with total genomic aberrations <100 Mb had a survival advantage compared to those with total genomic aberrations >100 Mb (Figure 4). Cluzeau et al.23 previously used a total genomic aberration number of 100 Mb to stratify prognostic groups among high-risk myelodysplastic syndrome patients treated with single agent azacitidine given as first-line therapy. Ganster et al.29 examined a cohort of very-high-risk myelodysplastic syndrome patients treated with azacitidine and lenalidomide, and showed that using a 200 Mb total genomic aberration cutoff would further stratify patients for overall survival. Taken together, a higher total genomic aberration number was associated with earlier death, although the total genomic aberrations cutoff differed between studies. Our data suggest there is a survival impact if total genomic aberrations is >100 MB and it could be incorporated into the next iteration of the IPPS score. However, before that a chromosome genomic array testing study with high total genomic aberrations may warrant prompt notification of the oncologist to allow closer monitoring/surveillance of the patient.
Our study further adds to the body of literature providing evidence that array testing improves diagnostic and prognostic yield in myelodysplastic syndrome. Volkert et al.26 showed that an additional 11% of myelodysplastic syndrome patients with normal karyotype had copy number aberrations when an array-based test was performed with conventional cytogenetics however, this study did not use a single-nucleotide polymorphism-containing array platform and therefore could not detect any copy neutral loss of heterozygosity. Several other studies have also shown that single-nucleotide polymorphism arrays enhance the diagnostic yield of myeloid stem cell disorders, from a range of 39–47% by metaphase chromosomal analysis to 54–74% by single-nucleotide polymorphism array.25, 30, 31 We demonstrated similar results in this study in that chromosome genomic array testing increased the abnormal detection rate for aberrancies from 57 to 73% when compared with karyotype. Of the subset with normal karyotype, chromosome genomic array testing detected abnormalities in 42% with both submicroscopic copy number aberration and somatic copy neutral loss of heterozygosity. Of the subset with unsuccessful karyotype testing, 100% showed informative chromosome genomic array testing results, underscoring the value of this assay in disease risk assessment.
Notably, in addition to the common abnormalities associated with myelodysplastic syndrome—including 5q deletions, monosomy 7, trisomy 8, and 20q deletions—gain of 1q and copy neutral loss of heterozygosity of several chromosome regions were prominent (Figure 1). In this cohort, copy neutral loss of heterozygosity of 9p was the most prevalent region afflicted by copy neutral loss of heterozygosity, followed closely by 11q, 17p, 4q, 11p, and 17q. Singh et al.32 reported frequent gain of 1q in patients with fibrosis and an association with advancing disease. Of the eight patients featuring 9p copy neutral loss of heterozygosity in our cohort, 50% showed myelofibrosis. Most of the copy neutral loss of heterozygosity regions we identified in this study are similar to those previously reported.33, 34, 35, 36, 37, 38, 39 These regions of copy neutral loss of heterozygosity are also areas where genes frequently containing myeloid disease-associated mutations are located, such as JAK2, CBL, TET2, EZH2, and TP53.
Correlating specific regions of copy neutral loss of heterozygosity with molecular and morphology data yielded intriguing results. A patient with 4q copy neutral loss of heterozygosity encompassing TET2 showed a splicing mutation with high allele frequency. One case with 9p copy neutral loss of heterozygosity also showed 11q copy neutral loss of heterozygosity encompassing the CBL gene and targeted gene panel next-generation sequencing showed a CBL splicing mutation. 9p copy neutral loss of heterozygosity was observed more frequently in patients with unilineage dysplasia as compared to 17p copy neutral loss of heterozygosity, which was more common in patients with multilineage dysplasia. This observation suggests that 9p copy neutral loss of heterozygosity might be limited to specific semi-committed hematopoietic cells in myelodysplastic syndrome pathogenesis, whereas 17p copy neutral loss of heterozygosity could occur in more pluripotent hematopoietic stem cells.40, 41 A recent study of myelodysplastic syndrome patients with 17p copy neutral loss of heterozygosity indicated an association with complex karyotype and homozygous TP53 mutations.42 Unfortunately, we could not sequence the two patients with copy neutral loss of heterozygosity of 17p in our cohort. Pairing the findings of copy neutral loss of heterozygosity with next-generation sequencing allowed us to understand the role these mutations may have, as mutations seen in the copy neutral loss of heterozygosity regions with a high allelic fraction by next-generation sequencing are supported to be homozygous and therefore more likely to have a role in the pathobiology of the disease.
Copy neutral loss of heterozygosity is an informative clonality marker and should be considered when making the diagnosis of myelodysplastic syndrome. Currently, these abnormalities can only be identified by chromosome genomic array testing or other single-nucleotide polymorphism-based array methodologies. Chromosome genomic array testing may be helpful in establishing the diagnosis thus improving risk stratification43 in samples with very low levels of morphologic abnormalities and no immunophenotypic abnormalities by flow cytometry. Our data indicate that the number of dysplastic lineages by morphology correlated with the total size of chromosome genomic array testing abnormalities. Our results have shown a significant survival advantage for myelodysplastic syndrome patients with lower total genomic aberrations, even when the analysis is performed among high-risk patient group. Conversely, an abnormal chromosome genomic array testing result is associated with adverse survival. Therefore, the presence of copy neutral loss of heterozygosity/copy number aberrations and total genomic aberration numbers obtained by chromosome genomic array testing analysis may provide clinically relevant prognostic information.
References
Ghaddar HM, Stass SA, Pierce S et al. Cytogenetic evolution following the transformation of myelodysplastic syndrome to acute myelogenous leukemia: implications on the overlap between the two diseases. Leukemia 1994;8:1649–1653.
Vardiman J, Hyjek E . World health organization classification, evaluation, and genetics of the myeloproliferative neoplasm variants. Hematology Am Soc Hematol Educ Program 2011;2011:250–256.
Vardiman JW . The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: an overview with emphasis on the myeloid neoplasms. Chem Biol Interact 2010;184:16–20.
Arber DA, Orazi A, Hasserjian R et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–2405.
Zeidan AM, Faltas B, Douglas Smith B et al. Myelodysplastic syndromes: what do hospitalists need to know? J Hosp Med 2013;8:351–357.
Glauser TA, Sagatys EM, Williamson JC et al. Current pathology practices in and barriers to MDS diagnosis. Leuk Res 2013;37:1656–1661.
Giagounidis A, Haase D . Morphology, cytogenetics and classification of MDS. Best Pract Res Clin Haematol 2013;26:337–353.
Deeg HJ, de Lima M . Hematopoietic stem cell transplantation for older patients with myelodysplastic syndromes. J Natl Compr Canc Netw 2013;11:1227–1233.
Deeg HJ, Bartenstein M . Allogeneic hematopoietic cell transplantation for myelodysplastic syndrome: current status. Arch Immunol Ther Exp (Warsz) 2012;60:31–41.
Haase D, Germing U, Schanz J et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 2007;110:4385–4395.
Gondek LP, Tiu R, O'Keefe CL et al. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008;111:1534–1542.
Arenillas L, Mallo M, Ramos F et al. Single nucleotide polymorphism array karyotyping: a diagnostic and prognostic tool in myelodysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes Cancer 2013;52:1167–1177.
Slovak ML, Smith DD, Bedell V et al. Assessing karyotype precision by microarray-based comparative genomic hybridization in the myelodysplastic/myeloproliferative syndromes. Mol Cytogenet 2010;3:23.
Kralovics R, Passamonti F, Buser AS et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779–1790.
Hagenkord JM, Monzon FA, Kash SF et al. Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of Affymetrix 10K2.0, 250 K Nsp, and SNP6.0 arrays. J Mol Diagn 2010;12:184–196.
Shaffer LG, Ballif BC, Schultz RA . The use of cytogenetic microarrays in myelodysplastic syndrome characterization. Methods Mol Biol 2013;973:69–85.
Mohamedali A, Gaken J, Twine NA et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood 2007;110:3365–3373.
Swerdlow SH . Myelodysplastic syndromes. In: Swerdlow SH E, Campo E, Harris N et al (eds). WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 4th edn. International Agency for Research on Cancer: Lyon, France, 2008, p 92.
Fang M, Storer B, Wood B et al. Prognostic impact of discordant results from cytogenetics and flow cytometry in patients with acute myeloid leukemia undergoing hematopoietic cell transplantation. Cancer 2012;118:2411–2419.
Wood BL . Ten-color immunophenotyping of hematopoietic cells. Curr Protoc Cytom 2005;Chapter 6 (Unit6):21.
Pritchard CC, Salipante SJ, Koehler K et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn 2014;16:56–67.
Shaffer LG, McGowan-Jordan J, Schmid M (eds). ISCN (2013): An International System for Human Cytogenetic Nomenclature (2013). Karger: Basel, Switzerland, 2013, 140 p.
Cluzeau T, Moreilhon C, Mounier N et al. Total genomic alteration as measured by SNP-array-based molecular karyotyping is predictive of overall survival in a cohort of MDS or AML patients treated with azacitidine. Blood Cancer J 2013;3:e155.
Paulsson K, Heidenblad M, Strombeck B et al. High-resolution genome-wide array-based comparative genome hybridization reveals cryptic chromosome changes in AML and MDS cases with trisomy 8 as the sole cytogenetic aberration. Leukemia 2006;20:840–846.
Tiu RV, Gondek LP, O'Keefe CL et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood 2011;117:4552–4560.
Volkert S, Haferlach T, Holzwarth J et al. Array CGH identifies copy number changes in 11% of 520 MDS patients with normal karyotype and uncovers prognostically relevant deletions. Leukemia 2016;30:257–260.
Dunbar AJ, Gondek LP, O'Keefe CL et al. 250 K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res 2008;68:10349–10357.
Bacher U, Haferlach T, Schnittger S et al. Investigation of 305 patients with myelodysplastic syndromes and 20q deletion for associated cytogenetic and molecular genetic lesions and their prognostic impact. Br J Haematol 2014;164:822–833.
Ganster C, Shirneshan K, Salinas-Riester G et al. Influence of total genomic alteration and chromosomal fragmentation on response to a combination of azacitidine and lenalidomide in a cohort of patients with very high risk MDS. Leuk Res 2015;39:1079–1087.
Bhatnagar B, Tiu RV, Gondek LP et al. Use of SNP-array-based karyotyping for cytogenetic prognostication in unclassified cases of myelodysplasia and associated overlap disorders. J Clin Oncol 2009;27:7016.
Huh J, Jung CW, Kim HJ et al. Different characteristics identified by single nucleotide polymorphism array analysis in leukemia suggest the need for different application strategies depending on disease category. Genes Chromosomes Cancer 2013;52:44–55.
Singh NR, Morris CM, Koleth M et al. Polyploidy in myelofibrosis: analysis by cytogenetic and SNP array indicates association with advancing disease. Mol Cytogenet 2013;6:59.
O'Keefe C, McDevitt MA, Maciejewski JP . Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood 2010;115:2731–2739.
Jankowska AM, Szpurka H, Tiu RV et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009;113:6403–6410.
Palomo L, Xicoy B, Garcia O et al. Impact of SNP array karyotyping on the diagnosis and the outcome of chronic myelomonocytic leukemia with low risk cytogenetic features or no metaphases. Am J Hematol 2016;91:185–192.
Merkerova MD, Bystricka D, Belickova M et al. From cryptic chromosomal lesions to pathologically relevant genes: integration of SNP-array with gene expression profiling in myelodysplastic syndrome with normal karyotype. Genes Chromosomes Cancer 2012;51:419–428.
Xu X, Johnson EB, Leverton L et al. The advantage of using SNP array in clinical testing for hematological malignancies—a comparative study of three genetic testing methods. Cancer Genet 2013;206:317–326.
Afable MG 2nd, Wlodarski M, Makishima H et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood 2011;117:6876–6884.
Heinrichs S, Kulkarni RV, Bueso-Ramos CE et al. Accurate detection of uniparental disomy and microdeletions by SNP array analysis in myelodysplastic syndromes with normal cytogenetics. Leukemia 2009;23:1605–1613.
Jones AV, Kreil S, Zoi K et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005;106:2162–2168.
Wang L, Swierczek SI, Lanikova L et al. The relationship of JAK2(V617F) and acquired UPD at chromosome 9p in polycythemia vera. Leukemia 2014;28:938–941.
Svobodova K, Zemanova Z, Lhotska H et al. Copy number neutral loss of heterozygosity at 17p and homozygous mutations of TP53 are associated with complex chromosomal aberrations in patients newly diagnosed with myelodysplastic syndromes. Leuk Res 2016;42:7–12.
Gronseth CM S, Scott B, CCS Yeung et al. Detection of cnLOH as a sole abnormality in the diagnosis of myelodysplastic syndrome. CAP Today 2016;82–86.
Acknowledgements
We thank the Seattle Cancer Care Alliance cytogenetics laboratory for the chromosome genomic array testing and karyotype data; UW Hematopathology Flow cytometry laboratory for the flow cytometry data; the UW Genomics and Molecular Pathology laboratory for next-generation sequencing and molecular data; and the Seattle Cancer Care Alliance Pathology Laboratory for the pathology slides and organization of the cases for review. This study was supported in part by a Pilot & Feasibility grant awarded to M. Fang under the Core Center of Excellence in Hematology Grant # P30 DK056465 and with support from the Cancer Center Support Grant # P30 CA015704 and Adult Leukemia Research Center Grant # P01 CA018029 as funded by the National Cancer Institute, National Institutes of Health, Bethesda, MD. CY received research funding from Gilead and MF received research funding from Affymetrix for projects that are unrelated to this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Yeung, C., McElhone, S., Chen, X. et al. Impact of copy neutral loss of heterozygosity and total genome aberrations on survival in myelodysplastic syndrome. Mod Pathol 31, 569–580 (2018). https://doi.org/10.1038/modpathol.2017.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.157
This article is cited by
-
Genomic variations in patients with myelodysplastic syndrome and karyotypes without numerical or structural changes
Scientific Reports (2021)
-
Clinical Applications of Chromosomal Microarray Testing in Myeloid Malignancies
Current Hematologic Malignancy Reports (2020)
