
Abstract
Monocytosis can develop during disease course in primary myelofibrosis simulating that seen in chronic myelomonocytic leukemia, and should not lead to disease reclassification. In contrast, at presentation, rare cases have clinical, morphologic, and molecular genetic features truly intermediate between primary myelofibrosis and chronic myelomonocytic leukemia. The taxonomy and natural history of these diseases are unclear. We identified cases which either: (1) fulfilled the 2008 World Health Organization criteria for primary myelofibrosis but had absolute monocytosis and, when available, chronic myelomonocytic leukemia-related mutations (ASXL1, SRSF2, TET2) or (2) fulfilled criteria of chronic myelomonocytic leukemia but had megakaryocytic proliferation and atypia, marrow fibrosis, and myeloproliferative-type driver mutations (JAK2, MPL, CALR). Patients with established primary myelofibrosis who developed monocytosis and those with chronic myelomonocytic leukemia with marrow fibrosis were excluded. By combining the pathology databases of two large institutions, six eligible cases were identified. Patients were predominantly male and elderly with monocytosis at diagnosis (average 17.5%/2.3 × 103/μl), organomegaly, primary myelofibrosis-like atypical megakaryocytes admixed with a variable number of chronic myelomonocytic leukemia-like hypolobated forms, variable myelodysplasia, marrow fibrosis and osteosclerosis. All had a normal karyotype and no myelodysplasia-associated cytogenetic abnormalities. Five of the patients in whom a more extensive molecular characterization was performed showed co-mutations involving JAK2 or MPL and ASXL1, SRSF2, TET2, NRAS, and/or KRAS. Disease progression has occurred in all and two have died. Rare patients present with features that overlap between primary myelofibrosis and chronic myelomonocytic leukemia and are thus difficult to classify based on current World Health Organization criteria. Biologically, these cases likely represent primary myelofibrosis with monocytosis, dysplasia, and secondary (non-driver) mutations at presentation. Alternatively, they may represent a true gray zone of neoplasms. Their clinical behavior appears aggressive and innovative therapeutic approaches may be beneficial in this particular subset.
Similar content being viewed by others
Main
Chronic myelomonocytic leukemia is a clonal hematopoietic stem cell neoplasm generally recognized as a chronic leukemia with hybrid features that include both myelodysplastic and myeloproliferative components. Chronic myelomonocytic leukemia is characterized by persistent monocytosis and dysplasia but also has features typically associated with myeloproliferative neoplasms including leukocytosis and/or splenomegaly. The simultaneous occurrence at initial presentation of both dysplasia and myeloproliferative features suggests that chronic myelomonocytic leukemia has elements of both myelodysplastic syndrome and myeloproliferative neoplasm and for this reason the current World Health Organization classification scheme identifies chronic myelomonocytic leukemia as a distinct entity with myelodysplastic/myeloproliferative overlap features.1 Chronic myelomonocytic leukemia is a heterogeneous myeloid neoplasm, however, with diverse clinical presentations, histologic features, and underlying molecular genetic abnormalities. Although variable, the clinical outcome of chronic myelomonocytic leukemia is generally poor.
Primary myelofibrosis is a clonal myeloproliferative neoplasm characterized by neoplastic granulocytic and megakaryocytic proliferation with erythroid hypoplasia. Patients with primary myelofibrosis typically present with anemia, leukocytosis and splenomegaly. Abnormal megakaryocytic morphology is a morphologic hallmark of the disease and is characterized by markedly atypical megakaryocytes present in tight clusters. Most megakaryocytes in primary myelofibrosis are enlarged in size although small forms can also be seen. The megakaryocytes generally have abnormal nuclear to cytoplasmic ratios and nuclear atypia in the form of abnormal chromatin clumping, segmentation, and hyperchromasia. Pronounced megakaryocytic atypia, in well-established disease, is usually seen in the context of marrow fibrosis and osteosclerosis. Somatic mutations in the signaling kinase JAK2 are seen in the majority of primary myelofibrosis (65%) although alternative mutations have been identified in the calcium storage protein calreticulin (CALR, 20%) and in the thrombopoietin receptor MPL (5%).2 The remaining primary myelofibrosis are "triple negative" for these mutually exclusive driver mutations.
Patients with primary myelofibrosis can develop monocytosis during the disease course, a feature that should not lead to reclassification of the myeloid neoplasm. Boiocchi et al3 reported 10 of 237 cases of primary myelofibrosis in which acquired monocytosis was identified during the disease course. In this report, monocytosis developed at a median interval of 42 months from initial diagnosis (range 1–180 months) and was associated with worsening cytopenias, increased circulating blasts and shorter survival. Clonal cytogenetic evolution was not seen in these patients. In a subset of these patients, follow-up bone marrow biopsies showed conversion from a primary myelofibrosis morphologic picture to one that would be consistent with chronic myelomonocytic leukemia. However, given the preexisting diagnosis of primary myelofibrosis, the initial disease classification was retained.
Distinct from the previous series reported by Boiocchi et al, we have seen rare cases of chronic myeloid neoplasms that have morphologic, clinical, and molecular genetic features truly intermediate between primary myelofibrosis and chronic myelomonocytic leukemia at the time of initial presentation. The precise classification and disease course of these neoplasms is uncertain. This study was undertaken to evaluate the characteristics of these cases.
Materials and methods
Case Selection
We retrospectively searched the pathology databases of the University of Miami and Weill Cornell Medical College/New York-Presbyterian Hospital in order to specifically identify cases provisionally diagnosed as “chronic myeloid neoplasm with monocytosis” at disease onset. We intentionally identified cases which were not precisely classified because of either: (1) the case fulfilled the 2008 World Health Organization criteria for primary myelofibrosis but had absolute monocytosis at presentation and, when available, myelomonocytic leukemia-associated mutations (ASXL1, SRSF2, and/or TET2) or (2) cases which fulfilled the 2008 World Health Organization criteria of chronic myelomonocytic leukemia but had megakaryocytic proliferation and atypia consistent with that of primary myelofibrosis, marrow fibrosis and, when available, one of the primary myelofibrosis-driver mutations (JAK2, MPL, CALR). Patients with established primary myelofibrosis who developed monocytosis and/or dysplasia, chronic myelomonocytic leukemia patients who developed marrow fibrosis and lacked other morphologic or clinical features of primary myelofibrosis, patients with increased blasts or possibly reactive monocytosis were excluded. Patients in whom initial diagnostic bone marrow biopsies were not available for our review were also excluded. By combining the pathology databases of two large institutions, six cases were identified which met eligibility criteria. The patient's initial bone marrow biopsy, aspirate smears, and peripheral blood smears were reviewed by at least one hematopathologist (JC, FV, JG, or AO; and all cases reviewed by AO). The presence of absolute monocytosis at initial diagnosis was confirmed by clinical medical record review. Relevant clinical parameters were retrieved from the clinical records of all patients.
Morphologic Assessment
Hematoxylin and eosin stain was used for evaluation of core biopsies and clot sections. Trichrome and reticulin stains were used to assess marrow fibrosis according to the European consensus criteria.1 Bone marrow aspirate smears and/or peripheral blood smears were stained with Wright–Giemsa and were evaluated in all cases.
Cytogenetic Assessment
Karyotyping
For bone marrow and peripheral blood specimens, multiple cultures were set up using Marrow Max and Chang media in a CO2 incubator and harvested after 24, 48, or 72 h of incubation at 37 °C, respectively. Cell cultures were treated with colcemid to arrest metaphase cells. Chromosome slides were prepared and stained using standard protocol for G banding. At least 20 metaphase cells were analyzed. Karyotypes were described and reported using an International System for Human Cytogenetic Nomenclature (ISCN 2016).4
Fluorescence in situ hybridization
Slides were prepared using bone marrow and/or peripheral blood specimens. Fluorescence in situ hybridization was performed using the protocols recommended by the manufacturers of the fluorescence in situ hybridization probes used. A dual-color probe was used to detect BCR-ABL1 gene fusion. A panel of probes was used for the 5q31, 5q33-q34, 7q31, and 20q12 chromosome regions as well as the centromeric region of chromosome 8 to detect the abnormalities commonly seen in myelodysplastic syndromes. A total of 200 interphase cells were analyzed for each sample with images captured and stored using Applied Imaging/Cytovision system. Final results were reported using the cutoffs established in the laboratory for each of the probes tested.
Mutation detection
Deoxyribonucleic acid was prepared from bone marrow aspirate and/or peripheral blood using a standard protocols established in the laboratories of the respective Universities at which the patients were diagnosed and treated (University of Miami, Weill Cornell University) and/or at a reference laboratory (Genoptix Laboratory). JAK2 mutation status was determined using allele-specific competitive blocker polymerase chain reaction assay. Sequencing based mutation detection was performed (Genoptix) on a panel of genes including ASXL1, BCOR, BRAF, CALR, CEBPA, CBL, CSF3R, DNMT3A, ETV6, EZH2, GATA2, GNAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NF1, NPM1, NRAS, PDGFRa, PDGFRb, PHF6, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, STAT3, STAT5b, TET2, TP53, U2AF1, WT1, and ZRSR2.
Results
Key clinical, histopathologic, and molecular genetic findings from the time of initial presentation of each patient are summarized in Table 1.
Patient 1
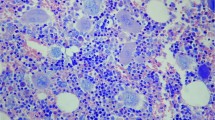
This patient is a 73-year-old male who initially presented with fatigue, flank pain and a 15-pound unintentional weight loss in June of 2013. He was found to have mild anemia and leukocytosis (hemoglobin 11 g/dl, white blood cell count 24.2 × 103/μl). His platelet count was 145 × 103/μl. He denied fevers, night sweats, and abdominal pain. Peripheral blood examination identified leukocytosis predominantly due to left-shifted granulocytosis with few circulating blasts (2%) (Figure 1a). Mild dysgranulopoiesis was present and was predominantly in the form of nuclear hyper- and hyposegmentation. Monocytes were increased comprising 4% of nucleated cells in the peripheral blood (absolute monocyte count 0.968 × 103/μl). JAK2 V617F mutation was detected in peripheral blood.

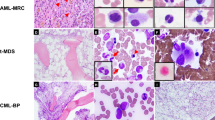
Peripheral blood smear shows granulocytosis with occasional dysplasia and circulating blasts (a). Bone marrow aspirate smears show granulocyte predominance with mild dysplastic features (cytoplasmic hypogranularity and nuclear segmentation abnormalities) (b, c). Bone marrow core biopsy shows granulocytic and megakaryocytic hyperplasia with increased, clustered and cytologically atypical megakaryocytes (with enlarged hyperchromatic nuclei), consistent with primary myelofibrosis-like morphology (d, e). Marrow fibrosis, grade myelofibrosis-2 (only Reticulin stain shown) (f). Images (a – c): Wright–Giemsa stain; (d, e): Hematoxylin and Eosin stain; (f): Reticulin stain. Images (a and c): 500 times magnification, (b and e): 400 times magnification, (d and f): 200 times magnification.
Bone marrow biopsy confirmed the presence of a myeloid neoplasm manifested by abnormally hypercellular marrow, >95%, owing to granulocyte predominant panmyeloid hyperplasia (Figure 1). Megakaryocytes were increased in number with abnormal clustering and significant cytologic atypia in the form of abnormal nuclear hypersegmentation, abnormal chromatin clumping, and hyperchromasia, consistent with primary myelofibrosis-like morphology. Associated marrow fibrosis was present, grade myelofibrosis-2. Aspirate smears were cellular and showed granulocyte predominant hematopoiesis with full maturation, blasts 1%. Dysgranulopoiesis and occasional dyserythropoiesis was present. Cytogenetic analysis identified a normal male karyotype in 20 metaphases. Fluorescence in situ hybridization for myelodysplasia-associated abnormalities was negative. Mutation analysis identified pathogenic alterations of JAK2, ASXL1, SRSF2, and TET2 genes (48, 42, 40, and 46% variant allele frequency, respectively). The findings were interpreted to be those of a chronic myeloid neoplasm with overall features most consistent with primary myelofibrosis but with monocytosis, dysplasia, and chronic myelomonocytic leukemia-associated mutations, suggesting overlap features with chronic myelomonocytic leukemia.
Over the subsequent 5 months the patient experienced increasing splenomegaly with flank pain, fatigue, and weight loss and was therefore started on ruxolitinib. Although he had initial improvement in his splenomegaly, his fatigue persisted. His white blood cell count progressively increased to 59.5 × 103/μl and his anemia worsened. Subsequent bone marrow biopsies demonstrated progression of marrow fibrosis and worsening of megakaryocytic atypia and granulocytic dysplasia. Blasts did not increase significantly over time. Stem cell transplantation was excluded based on the patient’s age and performance status. Following withdrawal of ruxolitinib, the patient was treated with an investigational agent (KB004, a humanized anti-EPhA3 receptor mono-clonal antibody) and maintained stable disease for almost a year, before progression of his disease once again. At this point, the patient was switched to an alternative investigational approach (alisertib, novel aurora kinase inhibitor) and achieved a working group response which he continues to maintain 40 months after initial presentation.
Patient 2
The patient is a 76-year-old male with a history of coronary artery disease and kidney tumor treated with cryoablation. One year later, he had progressive fatigue and was found to have anemia and circulating blasts. At presentation his complete blood count was as follows: white blood cell count 6.1 × 103/μl, hemoglobin 9.6 g/dl, hematocrit 28.5%, platelets 145 × 103/μl. Manual differential showed 6% blasts and 40% monocytes (absolute monocyte count of 2.44 × 103/μl) (Figure 2a). There was evidence of leukoerythroblastosis and dysgranulopoiesis in his peripheral blood. Abdominal imaging showed hepatosplenomegaly.

Peripheral blood smear shows absolute monocytosis and dysgranulopoiesis (a). Bone marrow core biopsy cellularity consisted predominantly of granulopoiesis and increased monocytic cells with scant erythropoiesis. Megakaryocytes were markedly increased and pleomorphic including many large forms with hyperchromatic, abnormally segmented, and occasionally bizarre nuclei (b). Marked marrow reticulin fibrosis was present (myelofibrosis-3) (c). Image (a): Wright–Giemsa stain: 1000 times magnification; (b): Hematoxylin and Eosin stain; (c): Reticulin stain. Images (b and c): 200 times magnification.
A bone marrow biopsy was markedly fibrotic (myelofibrosis-3) and hypercellular (>95%) (Figure 2). The marrow cellularity consisted predominantly of granulopoiesis and increased monocytic cells. The granulocytic series showed left-shifted but complete maturation. Blasts were not increased. Erythropoiesis was scant. Megakaryocytes were markedly increased and were pleomorphic with many large forms featuring hyperchromatic, abnormally segmented, and occasionally bizarre nuclei occasionally forming loose clusters.
Immunohistochemistry with CD14 showed ~30% monocytes and no increase in blasts. Bone marrow aspirate was aspicular and hemodilute with 7% blasts. Flow cytometry had 8% myeloblasts, which were CD34+, CD117+, HLA-DR+, CD13+, CD33+, CD14−, CD11b−, CD15 (partial), and CD7 (partial). Cytogenetic analysis showed a normal male karyotype and no abnormalities by fluorescence in situ hybridization. Polymerase chain reaction showed no evidence of JAK2 mutation. Further mutational analysis was not available (2013). The patient was treated with azacitidine but eventually died following progression to acute leukemia, 8 months after his initial presentation.
Patient 3
The patient is a 75-year-old woman with a history of Sjogren's syndrome, coronary artery disease, and common variable immunodeficiency, treated with intravenous immunoglobulin, and a 3-year history of leukocytosis (white blood cell count 12.4 × 103/μl with 57% neutrophils, 14.3% monocytes, absolute monocyte count 1.77 × 103/μl). She then presented with increasing fatigue. Complete blood count showed white blood cell count of 19.7 × 103/μl with myeloid left-shift, dysgranulopoiesis and 12.5% monocytes (absolute monocyte count, 2.46 × 103/μl) (Figure 3a). The remaining complete blood cell count values were within normal limits (hemoglobin 14.4 g/dl, hematocrit 42.1%, platelets 226 × 103/μl). Physical examination showed no evidence of organomegaly. Lactate dehydrogenase was increased at 315 U/L.

Peripheral blood smear shows dysgranulopoiesis. Absolute monocytosis was also present (not shown) (a). Bone marrow aspirate smear shows granulocytic predominance with mild granulocytic and erythroid dysplasia (b). Megakaryocytes are increased and range from small megakaryocytes with naked nuclei to large megakaryocytes with bulky, cloud-like nuclei. Prominent clustering was not seen. Immunohistochemical staining for CD14 demonstrated ~15% monocytes (inset) (c). Reticulin stain shows mildly increased reticulin fibrosis (no collagen fibrosis was noted and therefore marrow fibrosis graded myelofibrosis-1) (d). Images (a and b): Wright– Giemsa stain: 1000 times magnification; (c): Hematoxylin and Eosin stain: 200 times magnification; inset: CD14 immunohistochemistry stain: 400 times magnification; (d): Reticulin stain: 400 times magnification.
A bone marrow examination showed marked hypercellularity for age (95%) with prominent granulopoiesis (Figure 3). The granulocytic series showed complete maturation with a mild left-shift. Maturing erythropoiesis was very scant. Megakaryocytes were increased and ranged from small megakaryocytes with naked nuclei to large megakaryocytes with hypersegmented nuclei (Figure 3). Prominent clustering was not seen. Reticulin stain showed myelofibrosis-1 fibrosis. Bone marrow aspirate showed similar findings with myeloid: erythroid ratio of 4.1 and the presence of borderline myeloid and erythroid dysplasia (~10%). Sea-blue histiocytes were seen.
Immunohistochemical staining for CD14 demonstrated ~15% monocytes. There was no increase in blasts. Flow cytometry was non contributory. Cytogenetic analysis demonstrated a normal female karyotype. A 21-gene myeloid mutation sequencing panel revealed a JAK2 V617F mutation (60% variant allele frequency) and two pathogenic TET2 mutations (44 and 47% variant allele frequency).
The patient has been treated with ruxolitinib and has stable disease with persistent leukocytosis (12.9-30 × 103/μl) and absolute monocytosis (1.3–2.6 × 103/μl) after a follow-up of 54 months.
Patient 4
This patient is a 58-year-old male who initially presented with bilateral lower extremity edema. He was found to be profoundly anemic with leukocytosis and thrombocytopenia (hemoglobin of 12.4 g/dl, white blood cell count 17.9 × 103/μl, platelets 66 × 103/μl) and to have massive splenomegaly (palpable in the left iliac fossa). He complained of anergia and night sweats with a 40-pound unintentional weight loss over the previous year. Review of peripheral blood smear demonstrated leukocytosis due to left-shifted granulocytosis and monocytosis with granulocytic and monocytic dysplasia (Figure 4a). Absolute monocytosis was present (monocytes 8%, absolute monocyte count 1.43 × 103/μl). Bone marrow biopsy demonstrated an abnormally cellular marrow, 95%, due to granulocyte predominant panmyeloid hyperplasia with marrow fibrosis (myelofibrosis-3) (Figure 4). Dysplastic megakaryopoiesis was striking and included forms ranging from micromegakaryocytes with monolobated nuclei to large forms with polylobated and hyperchromatic nuclei. Abnormal megakaryocytic clustering was frequent. Aspirate smears confirmed granulocytic dysplasia in the form of nuclear segmentation abnormalities and cytoplasmic hypogranularity. Blasts comprised 3% of nucleated cells. Erythrocytes were decreased but were also often dysplastic. Storage iron was decreased (1+) and ring sideroblasts were not identified. Karyotype was that of a normal male in 20 metaphases and fluorescence in situ hybridization studies were negative for myelodysplasia-associated abnormalities and for BCR-ABL fusion. JAK2 V617F and JAK2 exon 12 mutation testing was negative. Mutation testing was positive for pathogenic alterations in MPL, ASXL1, SRSF2, TET2, TET2, and NRAS (variant allele frequencies of 44%, 43%, 46%, 48%, 42%, 25%, respectively). A genomic alteration of unclear significance was also identified in the EZH2 gene (51% variant allele frequency). It is likely that this mutation was also pathogenic given the high variant allele frequency and known association of EZH2 mutations and myeloid neoplasms.

Peripheral blood smear shows left-shifted and dysplastic granulocytosis and monocytosis (a). Aspirate smears confirmed granulocytic dysplasia (nuclear segmentation abnormalities and cytoplasmic hypogranularity) without increased blasts. Erythrocytes were decreased but were also often dysplastic (b). Bone marrow biopsy demonstrated granulocyte predominant panmyeloid hyperplasia with associated osteosclerosis (c). Dysplastic megakaryopoiesis was striking and included micromegakaryocytes with monolobated nuclei as well as large forms with polylobated and hyperchromatic nuclei. Abnormal megakaryocytic clustering was frequent (d, e). Marrow fibrosis was grade myelofibrosis-3 (trichrome stain not shown) (f). Images (a, b): Wright–Giemsa stain; (c, d, e): Hematoxylin and Eosin stain; (f): Reticulin stain. Images (a, b): 1000 times magnification; (c, d, f): 200 times magnification; (e): 400 times magnification.
The patient was started on ruxolitinib and experienced initial improvement of symptomatology with therapy, but his white blood cell count progressively increased to 43.5 × 103/μl with increasing monocytosis (absolute monocyte count 3.5 × 103/μl), and circulating blasts comprising 3% of nucleated cells. He remained anemic and thrombocytopenic over the next several months requiring intermittent transfusions. He received an allogeneic stem cell transplant but died subsequently from transplant related complications, 12 months after initial presentation.
Patient 5
The patient is a 74-year-old male who presented with an abnormal blood count noted during routine health maintenance. Complete blood count was as follows: white blood cell 15 × 103/μl, hemoglobin 13.6 g/dl, hematocrit 41.5%, MCV 87 fL, platelets 498 × 103/μl. Manual counts showed myeloid left-shift with granulocytic dysplasia, monocytosis (21%, absolute count 3.68 × 103/μl), eosinophilia (6%), and basophilia (4%) (Figure 5a). The patient had hepatosplenomegaly on physical exam.

Peripheral blood smear shows granulocytic dysplasia and monocytosis (a). Bone marrow biopsy demonstrats increased marrow cellularity with myeloid hyperplasia, full maturation, scant erythropoiesis, and increased megakaryocytes forming focal loose clusters with cytologic atypia including large forms and multinucleation (b). Image (a): Wright–Giemsa stain: 1000 times magnification; (b): Hematoxylin and Eosin stain: 200 times magnification.
A bone marrow biopsy demonstrated increased marrow cellularity (80%) with myeloid hyperplasia. Granulopoiesis showed full maturation. Erythropoiesis appeared megaloblastoid. Megakaryocytes were increased in number, formed focal loose clusters and had cytologic atypia including large forms and multinucleation (Figure 5). Reticulin and trichrome stains demonstrated myelofibrosis-2 fibrosis. Bone marrow aspirate confirmed a myeloid:erythroid ratio of 7.6 and evidence of borderline myeloid dysplasia.
Immunohistochemical staining with CD14 highlighted ~15% monocytes. Flow cytometry was non contributory. Cytogenetic analysis showed a normal male karyotype and no evidence of abnormalities by fluorescence in situ hybridization. 40-gene myeloid mutation sequencing panel showed JAK2 V617F (60% variant allele frequency), KRAS (33%), and TET2 (48%) mutations.
The patient has been managed expectantly and remains well despite a slow increase in his peripheral monocytosis 24 months after initial presentation.
Patient 6
This patient is a 63 year old man with history of colon cancer (post resection), psoriasis, and kidney stones. He presented with traumatic soft tissue laceration of his right leg. Complete blood count showed: white blood cell count 35.1 × 103/μl, hemoglobin 7 g/dl, hematocrit 20.9%, MCV 89.4 fL, platelets 263 × 103/μl. Manual differential count demonstrated myeloid left-shift including 4% blasts, 11% monocytes (absolute monocyte count 2.85 × 103/μl), and 2% basophils. There was evidence of leukoerythroblastosis. The patient had splenomegaly on physical exam.
Bone marrow examination demonstrated a markedly hypercellular (~100%) marrow with myeloid hyperplasia (Figure 6). Myeloid maturation was mildly left-shifted but was complete with numerous neutrophils seen. Erythroid maturation was decreased. Megakaryocytes were markedly increased in number and were large and hyperchromatic with numerous tight clusters. Myelofibrosis-3 marrow fibrosis with osteosclerosis was present. Bone marrow aspirate had scant spicules with myeloid to erythroid ratio of 6.1, 4% blasts, borderline dysgranulopoiesis and dyserythropoeisis, and monocytosis confirmed by non-specific esterase cytochemical stain (28%).

Bone marrow core biopsy demonstrates marked hypercellularity (~100%) with myeloid hyperplasia. Megakaryocytes are markedly increased in number, large and hyperchromatic with numerous tight clusters. Immunohistochemical staining with CD14 demonstrated ~30% monocytes in the core biopsy (inset) (a). Myelofibrosis-3 marrow fibrosis with osteosclerosis was present (b). Image a: Hematoxylin and Eosin stain including CD14 immunohistochemistry (inset); (b): Reticulin stain. Images (a and b): 200 times magnification.
Immunohistochemical staining with CD14 demonstrated ~30% monocytes in the core biopsy (Figure 6). Flow cytometry showed a population of CD34+, CD117+, CD13+, CD33+, CD123+ myeloid blasts (1.5% of analyzed cells). Monocytes were increased and had aberrant expression of CD56. Cytogenetic analysis had a normal male karyotype and no evidence of BCR-ABL1 gene rearrangement by fluorescence in situ hybridization. 21-gene myeloid mutation sequencing panel showed JAK2 V617F, and TET2 mutations (variant allele frequency not available). The patient was lost to follow-up 2 months after the initial diagnosis.
Summary of the cases with “gray zone” features
Patients in this cohort were predominantly male (83%) and elderly (average 69.8 years, range 58–76). At disease onset, patients had leukocytosis and absolute monocytosis (average 17.5%/2.3 × 103/μl, range 4–40%/0.97–3.7 × 103/μl), organomegaly (83%), and all had primary myelofibrosis-like atypical megakaryocytes admixed with a variable number of chronic myelomonocytic leukemia-like hypolobated forms, variable degrees of myelodysplasia, marrow fibrosis and osteosclerosis. All patients had normal karyotypes and no myelodysplasia-associated cytogenetic abnormalities. Five of the patients in whom molecular characterization was performed showed co-mutations involving JAK2 or MPL with a variable combination of TET2, ASXL1, SRSF2, NRAS, KRAS, and/or EZH2 mutations (See Table 1). Three patients were treated with ruxolitinib, one followed by a series of two investigational agents, one with azacitidine, one is being observed and one was lost to follow-up. Four out of five (80%) patients with clinical follow-up showed disease progression, and two have died (one of disease progression to acute leukemia, one from complications of transplant).
Discussion
A diagnosis of chronic myelomonocytic leukemia is made when there is persistent and otherwise unexplained absolute monocytosis in peripheral blood (monocytes ≥1 × 103/μl, monocytes ≥10% of leukocytes), BCR-ABL negativity, absence of PDGFRA and PDGFRB rearrangements, and <20% blasts.5 Although dysplasia is typically present in chronic myelomonocytic leukemia, the diagnosis can be made in the absence of dysplasia if the other diagnostic criteria are met and the monocytosis is persistent and otherwise unexplained, or if a clonal cytogenetic or molecular abnormality is identified. Dysplasia, particularly of granulocytes, while often present may be less pronounced in patients with higher white blood cell counts (proliferative type of chronic myelomonocytic leukemia).5 Bone marrow biopsies are hypercellular due to granulocytic hyperplasia with associated monocytic hyperplasia. Erythroid precursors are not typically increased in number but often demonstrate dysplasia in aspirate smears. Megakaryopoiesis is variable. Megakaryocytes may be normal in morphologic appearance but are more often dysplastic. Dysmegakaryopoiesis in chronic myelomonocytic leukemia is characterized by the presence of dwarf megakaryocytes and/or megakaryocytes with hypolobated or abnormally lobulated nuclei. Such dysmegakaryopoiesis is found in 75% of chronic myelomonocytic leukemia cases.6
Based on the diversity of clinical presentation and pathologic findings, chronic myelomonocytic leukemia was initially divided into two subsets in the French–American–British classification scheme based on peripheral white blood cell count: myelodysplastic-chronic myelomonocytic leukemia (white blood cell count <13 × 106/μl) and myeloproliferative-chronic myelomonocytic leukemia (white blood cell count ≥13 × 106/μl). This division of chronic myelomonocytic leukemia into myelodysplastic and myeloproliferative types has subsequently been shown to be of prognostic significance and to correlate with differences in RAS/ MAPK signaling pathways, thus the revised 2016 World Health Organization classification will maintain the classification of chronic myelomonocytic leukemia into myelodysplastic and myeloproliferative types.5 In addition to lower white blood cell count, myelodysplastic-chronic myelomonocytic leukemia usually has absent splenomegaly and more pronounced dysplasia. Splenomegaly is seen more commonly in myeloproliferative-chronic myelomonocytic leukemia.
Owing to the low incidence of chronic myelomonocytic leukemia, its relatively new acceptance as a distinct entity, and the heterogeneity of its clinical presentation and histopathologic features, the majority of studies regarding the biology and outcome of chronic myelomonocytic leukemia have not been performed as isolated studies of chronic myelomonocytic leukemia. Rather, most investigations into the biology of chronic myelomonocytic leukemia have been performed in the context of myelodysplasia-focused clinical trials in which chronic myelomonocytic leukemia is included as a subtype of myelodysplastic syndrome. Increasing availability of cytogenetic and gene sequencing assays, however, have shown that while most cases of chronic myelomonocytic leukemia do have cytogenetic and/genetic abnormalities, these abnormalities are heterogeneous and none are specific for chronic myelomonocytic leukemia. The clinical and histologic variability of chronic myelomonocytic leukemia is likely the result of this underlying diverse genetic landscape.
Clonal cytogenetic abnormalities are seen in ~30% of conventional chronic myelomonocytic leukemia patients, whereas molecular abnormalities are seen in >90%.7, 8 Several different chromosomal translocations have been reported in chronic myelomonocytic leukemia, each being individually rare. Molecular abnormalities reported in chronic myelomonocytic leukemia, which are much more common, include mutations involving epigenetic regulator genes (TET2 most common, 60%), chromatin regulation, and histone modification (ASXL1 most common, 40%), splicing machinery (SRSF2 most common, 50%), and those involving signal transduction and receptor tyrosine kinase pathways (JAK2, 15% and RAS, 30%). Taken together, the vast majority of chronic myelomonocytic leukemia will have TET2, SRSF2, ASXL1, and/or JAK2/ RAS gene mutations, none of which is specific for identification of chronic myelomonocytic leukemia among other myeloid neoplasms. While the prognostic impact of some of these more common mutations is controversial, ASXL1 mutations have been shown to negatively impact overall survival in chronic myelomonocytic leukemia.8 TET2 mutations, on the other hand, have not been shown to be independently predictive of overall survival or leukemia-free survival.7
Different than chronic myelomonocytic leukemia, primary myelofibrosis is characterized by neoplastic granulocytic and megakaryocytic proliferation with erythroid hypoplasia and a variable degree of marrow fibrosis. Abnormal megakaryocytic morphology is the hallmark of primary myelofibrosis and is characterized by abnormally clustered and enlarged megakaryocytes with hypersegmented nuclear lobes. In addition to primary myelofibrosis-type megakaryocytic atypia, presence of fibrosis and/or granulocytic hyperplasia and erythroid hypoplasia, additional major diagnostic criteria of primary myelofibrosis include absence of BCR-ABL1 fusion gene and presence of a so-called "MPN driver" mutation, which include mutation of JAK2, MPL, or CALR. Although one of these driver mutations are identified in the vast majority of primary myelofibrosis, their presence is not specific for primary myelofibrosis as they are also seen in the other Philadelphia-negative chronic myeloproliferative neoplasms. In addition to the presence of a driver mutation, primary myelofibrosis can also acquire additional mutations, the majority of which are loss of function mutations and thus it is believed that the mutated genes are myeloid tumor suppressor genes. Secondary genes most commonly mutated in primary myelofibrosis include TET2, DNMT3A, EZH2, ASXL1, SRSF2, and U2AF1.9 Loss of function mutations in genes involved in epigenetic regulation (TET2, ASXL1), and mutations in regulatory and splicing genes whose mutation cooperates with the MPN driver mutation are thought to play a key role in the pathogenesis of primary myelofibrosis. These additional, presumably acquired and/or secondary, mutations are common in primary myelofibrosis. Importantly, however, none of these secondary mutations are restricted to primary myelofibrosis or even myeloproliferative neoplasms and they are, in fact, significantly more common in chronic myelomonocytic leukemia, myelodysplastic syndromes, and acute myeloid leukemias.
Li et al recently described the identification of additional, non-driver, mutations in 96% of primary myelofibrosis patients in their cohort. Most frequent mutations in that study affected ASXL1 (33%), U2AF1 (22%), TET2 (16%), FAT1 (16%), SETBP1 (13%), and SRSF2 (9%), among others.10 A review by Vainchenker et al also reiterated that mutations in epigenetic regulators such as TET2 and DNMT3A are involved in the pathogenesis of primary myelofibrosis and in fact may precede the acquisition of JAK2 or other driver mutations.9 The frequent presence of the same non-driver mutations in other myeloid tumors (myelodysplastic syndromes, acute myeloid leukemias), supports the notion that these secondary mutations are important in affecting phenotypic features of the disease (histopathologic and clinical) and in disease progression, but are not necessarily disease-defining. Given that none of these mutations are specific for any myeloid neoplasm, their identification, whereas prognostically significant, cannot be used for disease classification. These findings support that fidelity of myeloid mutations is not absolute among disease categories. For example, the occurrence of driver mutations classically associated with myeloproliferative neoplasms, such as JAK2 mutation, can occur in other disease categories such as myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms including conventional chronic myelomonocytic leukemia. Likewise, myeloproliferative neoplasms can acquire myelodysplasia-related mutations, particularly at the time of disease progression. Although these acquired mutations may affect disease phenotype and be of prognostic significance, they should not be used in isolation to establish or change disease classification. Because of the nonspecificity of the myeloid mutations, cases of myeloid neoplasms where clinical and histologic findings are consistent with a given myeloid tumor should be classified as such and following the World Health Organization scheme. This is distinct from the cases presented here where the clinical, histologic, and molecular genetic features were all indeterminate between those of primary myelofibrosis and chronic myelomonocytic leukemia at the time of initial presentation, complicating disease classification independent of overlap mutational status.
As with many chronic myeloid neoplasms, patients with primary myelofibrosis can develop monocytosis and dysplasia during disease course, a feature which has been shown to be associated with progressive disease and shorter survival.3 Development of monocytosis and dysplasia in patients with previously established primary myelofibrosis does not pose a diagnostic challenge. However, if patients with presumably undiagnosed primary myelofibrosis present initially with monocytosis and dysplasia, these cases may be misclassified as chronic myelomonocytic leukemia. Indeed chronic myelomonocytic leukemia can be morphologically and clinically indistinguishable from primary myelofibrosis with monocytosis and dysplasia and can have the same molecular genetic abnormalities, including JAK2 mutations, additionally complicating disease classification.
Distinct from the development of acquired monocytosis and dysplasia during disease course in primary myelofibrosis, we have seen rare cases of chronic myeloid neoplasms that have clinical, morphologic and, when available, molecular genetic features truly intermediate between primary myelofibrosis and chronic myelomonocytic leukemia at the time of initial presentation. These cases are difficult to reproducibly classify and the natural history of such neoplasms is not known. Specifically, each case presented meets diagnostic criteria for primary myelofibrosis based on presence of major diagnostic criterion (primary myelofibrosis-like megakaryocytic atypia in the context of marrow fibrosis, absence of evidence of chronic myelogenous leukemia or other myeloproliferative or myelodysplastic neoplasm, presence of JAK2, MPL, or other mutation serving as a clonal marker) and variable combinations of at least one of the minor diagnostic criteria (anemia, leukoerythroblastosis, leukocytosis, splenomegaly). However, in addition to meeting these established criteria for primary myelofibrosis, each of the presented cases had additional features (leukocytosis that included absolute monocytosis, variable degrees of granulocytic and erythroid dysplasia as well as a variable number of small micromegakaryocytes) that are not typical of primary myelofibrosis and may suggest an alternative diagnosis of chronic myelomonocytic leukemia as defined by the World Health Organization 2008 Classification.1 The 2016 revised World Health Organization classification update has added a minimum level of monocytosis to the diagnostic criteria of chronic myelomonocytic leukemia which requires monocytes to account for ≥10% of the white blood cell differential count.5 Adding this criterion will have the overall effect of decreasing the number of cases meeting requirements for the diagnosis of chronic myelomonocytic leukemia. In our case series, for example, three of our cases which met criteria for chronic myelomonocytic leukemia according to the 2008 scheme do not meet criteria for this disease according to the 2016 scheme.
Monocytosis in all patients was persistent over time and was found in association with genetic mutations that are common in chronic myelomonocytic leukemia including TET2, ASXL1, SRSF2, NRAS, and/ or KRAS mutations in five of six patients (molecular information not available in one patient). Progression in the form of increasing leukocytosis with persistent monocytosis was seen in 4/5 (80%) patients with follow-up intervals ranging from 8 to 54 months. Two of these patients have died of disease, one after progression to acute leukemia (8 months after presentation, patient 2) and one after complications of bone marrow transplant performed for progressive disease (12 months after initial presentation, patient 4). One patient was lost to follow-up (patient 6).
It is unclear at this time whether these neoplasms truly represent overlap cases that have hybrid features of both primary myelofibrosis and chronic myelomonocytic leukemia or whether they represent primary myelofibrosis presenting initially with monocytosis, dysplasia, and “chronic myelomonocytic leukemia-associated” genetic abnormalities. In the latter circumstance, the secondary (non-driver) mutations likely play a role in determining disease phenotype and promoting progression, but may not necessarily be indicative of a “chronic myelomonocytic leukemia-associated” genetic phenotype. Elliot et al has reported previously the presence of absolute monocytosis at diagnosis in a subset (15.5%) of patients in their primary myelofibrosis cohort, and demonstrated that monocytosis at diagnosis was an adverse prognostic factor for survival in younger patients with primary myelofibrosis.11 In this previous report, cases meeting diagnostic criteria of chronic myelomonocytic leukemia were specifically excluded. Whether those cases of primary myelofibrosis with monocytosis at disease onset had morphologic or molecular genetic features overlapping with those of chronic myelomonocytic leukemia (dysplasia, chronic myelomonocytic leukemia-like micromegakaryocytes, chronic myelomonocytic leukemia-associated mutations) was not defined.
Given the well-documented incidence of acquired monocytosis and dysplasia in the context of primary myelofibrosis disease course and the striking primary myelofibrosis-like morphology seen in all cases in our cohort, we believe these may be examples of primary myelofibrosis initially presenting with monocytosis and myeloid dysplasia. If this is the case, then such presentation in this cohort was associated with the presence of additional non-driver mutations at the time of initial diagnosis including those of TET2, ASXL1, SRSF2, KRAS, NRAS, and EZH2. Given that the variant allele frequency of the myeloproliferative neoplasm-associated driver mutations (JAK2) was higher than that of the non-driver mutations in two of these cases, it is possible that the myeloproliferative neoplasm-associated driver mutation was an earlier genetic lesion that was followed by acquisition of the non-driver mutations. However, one of our cases (patient 4) had mutations of TET2 and SRSF2 present at variant allele frequency slightly higher than that of the associated driver (MPL) mutation, thus it is possible that these non-driver mutations were acquired first, as has been previously reported in primary myelofibrosis.10
Both primary myelofibrosis and chronic myelomonocytic leukemia are invariably fatal diseases unless treated with allogeneic hematopoietic cell transplantation. Some cases are indolent, but most patients will die from the disease. Hematopoietic cell transplantation is either not available or unsuccessful in most patients, the majority of which are older adults. Gray zone cases are particularly difficulty to manage, but standard therapy typically consists of, as in our cases, either primary myelofibrosis-directed (ruxolitinib) or chronic myelomonocytic-directed (hypomethylating agent) therapy. Such patients, as in our patient 1, are ideally treated on a clinical trial. However, many may not qualify under current diagnostic criteria. Recent data suggest that JAK/STAT pathway inhibitors (such as ruxolitinib) may be effective in chronic myelomonocytic leukemia, independent of JAK mutation status,12, 13 and it stands to reason that JAK inhibitors could be particularly effective in gray zone cases. Combinations of JAK inhibitors with MEK inhibitors (targeting RAS), hypomethylating agents, or other chronic myelomonocytic leukemia-directed therapy are promising novel combinations, and should be pursued at specialized centers, particularly in gray zone cases.14
Additional cases similar to those presented herein will need to be identified and studied so that the nature and thus the classification of these neoplasms can be better understood. The increasing precision of the World Health Organization classification scheme coupled with the now widespread availability of molecular genetic testing will help with identification of such cases. Such studies will likely lead to enhancements in reproducible classification of these myeloid neoplasms and also in the development of innovative therapeutic approaches, which are needed for these patients.
References
Orazi A, Bennett JM, Germing U et alChronic myelomonocytic leukaemia. In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, 2008;76–79.
Rozovski U, Verstovsek S, Manshouri T et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and MPL mutation status for patients with primary myelofibrosis. Haematologica 2017;102:79–84.
Boiocchi L, Espinal-Witter R, Geyer JT et al. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod Pathol 2013;26:204–212.
McGowan-Jordan J, Simons A, Schmid M . Iscn 2016: An International System for Human Cytogenomic Nomenclature 2016: S. Karger AG 2016.
Arber DA, Orazi A, Hasserjian R et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–2405.
Orazi A, Chiu R, O'Malley DP et al. Chronic myelomonocytic leukemia: the role of bone marrow biopsy immunohistology. Mod Pathol 2006;19:1536–1545.
Patnaik MM, Itzykson R, Lasho TL et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia 2014;28:2206–2212.
Nazha A, Prebet T, Gore S et al. Chronic myelomonocytic leukemia: are we finally solving the identity crisis? Blood Rev 2016;30:381–388.
Vainchenker W, Kralovics R . Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017;129:667–679.
Li B, Zhang L, Bai J et al. Non-Driver mutations profile identified by a 206-Gene NGS panel in patients with primary myelofibrosis and post-polycythaemic/essential thromocythaemia myelofibrosis in a single center from China. Blood 2016;128:1942.
Elliott MA, Verstovsek S, Dingli D et al. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk Res 2007;31:1503–1509.
Padron E, Dezern A, Andrade-Campos M et al. A multi-institution phase I trial of ruxolitinib in patients with Chronic myelomonocytic leukemia (CMML). Clin Cancer Res 2016;22:3746–3754.
Francke S, Mies A, Meggendorfer M et al. Disease-modifying activity of ruxolitinib in a patient with JAK2-negative CMML-2. Leuk Lymphoma 2017;58:1271–1272.
Kong G, Wunderlich M, Yang D et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest 2014;124:2762–2773.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Chapman, J., Geyer, J., Khanlari, M. et al. Myeloid neoplasms with features intermediate between primary myelofibrosis and chronic myelomonocytic leukemia. Mod Pathol 31, 429–441 (2018). https://doi.org/10.1038/modpathol.2017.148
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.148
This article is cited by
-
Advances in myelodysplastic/myeloproliferative neoplasms
Virchows Archiv (2023)
-
Clinicopathologic spectrum of myeloid neoplasms with concurrent myeloproliferative neoplasm driver mutations and SRSF2 mutations
Modern Pathology (2022)
-
Myeloproliferative Neoplasms with Monocytosis
Current Hematologic Malignancy Reports (2022)
-
Laboratory Evaluation and Pathological Workup of Neoplastic Monocytosis — Chronic Myelomonocytic Leukemia and Beyond
Current Hematologic Malignancy Reports (2021)
-
Update on the pathologic diagnosis of chronic myelomonocytic leukemia
Modern Pathology (2019)
