
Abstract
Neuroendocrine breast carcinomas represent a rare subtype of breast cancer. Their definition, prevalence, and prognosis remain controversial in the literature. The 2012 WHO classification of breast cancer categorizes neuroendocrine carcinomas into three morphologically distinct subtypes: well-differentiated neuroendocrine tumors, poorly differentiated neuroendocrine carcinomas, and invasive breast carcinomas with neuroendocrine differentiation. We aimed to gain insight into the clinical, morphologic, phenotypic, and molecular features of 47 neuroendocrine breast carcinomas. Targeted next-generation sequencing by an AmpliSeq 22 cancer gene hotspot panel and the Prosigna assay were performed on 42/47 and 35/47 cases, respectively. Average age at diagnosis was 69 years. All tumors were estrogen receptor-positive and the large majority expressed progesterone receptor (89%), GATA3 (98%), FOXA1 (96%), and CK8/18 (98%). There was an almost equal distribution of luminal A (52%) and B (48%) carcinomas. Almost half of the cohort (49%) displayed a high risk of recurrence score with the Prosigna test. Patients with a neuroendocrine carcinoma had a shorter disease-free survival compared with those affected by carcinomas of no special type matched for age, size, grade, and estrogen receptor status. No significant differences were observed in terms of overall survival. Stratification of neuroendocrine carcinomas using the 2012 WHO criteria did not reveal statistically significant differences among the distinct categories (well-differentiated neuroendocrine tumors, poorly differentiated neuroendocrine carcinomas, and invasive breast carcinomas with neuroendocrine differentiation), in terms of either progression-free or overall survival. Our targeted sequencing analysis found three cases (7%) harboring a PIK3CA mutation, and in three other cases (7%) TP53 mutations were detected. This study showed that neuroendocrine breast carcinoma is a distinct subtype of luminal carcinoma with a low rate of PIK3CA mutations and with an aggressive clinical behavior. An accurate identification of neuroendocrine differentiation may be useful to better tailor patient adjuvant therapy within luminal carcinomas.
Similar content being viewed by others


Main
Neuroendocrine breast carcinoma is a rare special histologic type of breast cancer that has similar morphologic and phenotypic characteristics to digestive and pulmonary neuroendocrine tumors1 but also shows a certain degree of heterogeneity, including some features often indistinguishable from those of invasive carcinomas of no special type.2 They also exceptionally present with a functional syndrome.3
In the breast, Feyrter and Hartmann recognized neuroendocrine carcinomas in 1963.1 Using Grimelius silver-staining techniques, they suggested the endocrine nature of some primary breast carcinomas. In 2003, the WHO classification of tumors of the breast and female genital organs defined neuroendocrine carcinomas as mammary carcinomas showing more than 50% of neoplastic cells expressing neuroendocrine markers by immunohistochemistry (synaptophysin, chromogranin A).4 The revised 2012 WHO classification5 includes three categories of neuroendocrine carcinomas: (i) well-differentiated carcinoma (densely cellular, solid nests, or trabeculae of cells that vary from spindle to plasmocytoid and large clear cells separated by fine vascular stroma); (ii) poorly differentiated/small cell carcinoma (morphologically indistinguishable from its counterpart in the lung); (iii) carcinoma of no special type or for instance mucinous hypercellular variant invasive breast carcinoma with neuroendocrine differentiation demonstrated on immunohistochemistry. In this new edition, the definition of neuroendocrine carcinomas is unclear as there is no minimal percentage of tumor cell-expressing neuroendocrine markers required for a diagnosis of neuroendocrine breast carcinoma.
Changes in classifications between 2003 and 2012 affect the comparison between different studies in the literature as different terminologies have been used. Some followed their own classification before 2003, or later, finding the WHO classification inadequate.6, 7, 8
The lack of uniformity in the definition and classification of neuroendocrine carcinomas hampers an exact estimate of the prevalence of these tumors, ranging from 0.1 to 15% depending on the series.9, 10, 11, 12, 13 This may affect also the controversial data on the prognostic implication of neuroendocrine differentiation in breast cancer. Some have reported a better prognosis in neuroendocrine carcinomas than in patients with invasive carcinomas of no special type,9, 11, 14, 15 whereas more recently a worse prognosis has been observed in larger series of neuroendocrine carcinomas as compared with carcinomas of no special type.9, 11, 14, 15
Molecular data on neuroendocrine carcinomas are scarce. Weigelt et al16 showed that these are luminal tumors with transcriptomic profiles similar to type B mucinous carcinomas (hypercellular variant). Two studies have investigated the mutational profiling of small cohorts of neuroendocrine carcinomas by using targeted sequencing panels17, 18 and identified recurrent mutations affecting PIK3CA,17, 18 the FGFR family,17 and chromatin-remodeling genes.18
The aims of our study were (i) to characterize the molecular and phenotypic aspects of neuroendocrine carcinomas of the breast, (ii) to assess differences between the three main morphologic subtypes of neuroendocrine carcinomas, and (iii) to assess the prognostic impact of the distinct categories of this rare subtype.
Materials and methods
Cases
A series of 47 neuroendocrine carcinomas were retrieved from the files of Institut Curie, France, and The University Hospital of Geneva, Switzerland by searching in the pathology files of the authors’ institutions using the following terms: ‘Neuroendocrine; Solid papillary; Mucinous; Chromogranin; Synaptophysin’ by text recognition system within the informatics medical files of the patients.
Patients’ identification was anonymized before analysis. Representative H&E (hematoxylin and eosin) slides were reviewed by three pathologists (ML, EM, and AVS) at a multiheaded microscope to confirm the diagnosis and to categorize the cases into one of the morphologic subtypes: well-differentiated, poorly differentiated/small cell carcinoma, and invasive breast carcinoma with neuroendocrine differentiation according to the histologic criteria described above. For each tumor, histologic grade was assessed using the Nottingham grading system.19 The presence of associated ductal carcinoma in situ, lymphovascular invasion, necrosis, and node metastasis was also recorded. To assess the potential impact of neuroendocrine differentiation on overall survival and disease-free survival, we strictly matched each of our cases with patients affected by invasive carcinomas of no special type, based on age at diagnosis, histologic tumor size, histologic grade, and hormone receptor status. A maximum of two controls for each neuroendocrine carcinoma was included.
This series of matched invasive carcinomas of no special type that served as a control group was retrieved from the files of Institut Curie.
Samples and Immunohistochemistry
Following slide review, the most representative paraffin block was selected for immunohistochemical analysis. Details about antibodies, clones, dilutions, and antigen retrieval methods used for each single antibody are reported in Supplementary Table 1. For each case, six immunohistochemical stainings were performed on full sections to confirm the diagnosis of neuroendocrine carcinoma and evaluate prognostic factors: estrogen receptor, progesterone receptor, HER2, Ki67, chromogranin A, and synaptophysin.
Furthermore, tissue microarrays were constructed from paraffin blocks with duplicate tumor cores. Normal peritumoral tissue was included in the tissue microarrays as a control. Immunohistochemistry was performed on 3-μm-thick tissue microarray sections, using a panel of antibodies against androgen receptor, Forkhead Box A1 (FOXA1), GATA-binding protein 3 (GATA3), Cytokeratin (CK) 8/18, CK14, Epidermal Growth Factor Receptor (EGFR), Thyroid Transcription factor 1 (TTF1), and Caudal type Homeobox 2 (CDX2). Positive and negative controls were included in each experiment.
FOXA1, GATA3, AR, and CDX2 expression levels were also investigated in the control group of invasive carcinomas of no special type matched with neuroendocrine carcinomas based on age at diagnosis, histologic tumor size, histologic grade, and hormone receptor status (available samples: n=30).
The stainings were interpreted semiquantitatively by a pathologist (ML), who was blinded to genomic data. Inclusion criteria for a diagnosis of neuroendocrine carcinoma were the expression of at least one neuroendocrine marker (synaptophysin or chromogranin A) in at least 50% of tumor cells. The expression of estrogen and progesterone receptors was interpreted following ASCO recommendations.20 Ki67, chromogranin A, synaptophysin, androgen receptor, CK14, EGFR, CDX2, and TTF1 were reported as a percentage of positive tumor cells. For GATA3, FOXA1, androgen receptor, and CK8/18, percentage of staining was categorized as '0' if there was no nuclear expression (or cytoplasmic for CK8/18), '1' for up to 10% positive tumor nuclei, '2' for 11–20%, and so on till a maximum score of '10.' Intensity was scored as '1+,' '2+,' and '3+' for weak, moderate, and strong staining, respectively. Percentage and intensity of nuclear expression were multiplied to generate a score (Score=Percentage × Intensity).
The scoring system updated according to the 2013 ASCO guidelines21 was used for HER2 assessment and fluorescence in situ hybridization was performed in case of an equivocal score 2+ result.
Tumors were then classified into molecular subtypes described by Goldhirsh et al22 into luminal A, luminal B, HER2-enriched, and basal-like, according to their hormone receptor status, HER2 status, and proliferation assessed by the Ki67 index.
Macrodissection and DNA Extraction
DNA extraction was possible for 45 cases (for two biopsy cases, the representative tumor material in the paraffin blocks was scarce). Enrichment in tumor cell content was performed with a punch biopsy in order to ensure a percentage of tumor cells greater than 30%. Five samples showed a tumor cellularity of 30%, whereas all of the others displayed a tumor cell content ranging from 40 to 80%. DNA was then extracted using NucleoSpin 8 tissue kit (Macherey-Nagel, Germany). Double-stranded DNA concentration was measured by fluorimetric method, using Qubit dsDNA HS Assay (Life Technology). DNA quality was assessed with qPCR EGFR using Light Cycler 480 technology.
Out of 45 microdissected carcinomas, 42 yielded DNA of sufficient quantity and quality for sequencing.
Targeted Next-Generation Sequencing
Forty-two cases were tested on the Ion PGM (Personal Genome Machine) Sequencer (Life Technologies).
We used an Ion AmpliSeq Cancer Panel that covers mutational hotspots of 22 genes: KRAS, EGFR,BRAF,PIK3CA,AKT1,ERBB2,PTEN,NRAS,STK11,MAP2K1,ALK,DDR2,CTNNB1,MET,TP53,SMAD4, FBXW7,FGFR3,NOTCH1,ERBB4,FGFR1,FGFR2.
In brief, 10 ng of DNA was amplified by PCR using Ion Ampliseq Library Kit 2.0. The 90 multiplexed amplicons generated were then ligated to adapters from the Ion Xpress Barcode Adapters Kits according to the manufacturer’s instructions (Ion Torrent).
Multiplexed barcoded libraries were amplified by emulsion PCR on Ion Sphere particles (Ion PGM Template OT2 200 Kit). Sequencing was performed on a PGM sequencer (ION Torrent) using the PGM Sequencing 200 Kit V2 according to the manufacturer’s instructions. Torrent Suite software version 3.4.2 was used to parse barcode reads, to align reads to the reference genome, and to generate run metrics.
Comparisons with Invasive Breast Carcinomas from the TCGA and the METABRIC Data Sets
To compare the mutational frequencies of neuroendocrine carcinomas to those of estrogen receptor-positive and PAM50-defined luminal A and B carcinomas, we interrogated the TCGA and METABRIC data sets. For the TCGA data set23 we retrieved the clinicopathologic data from the UCSC Xena Browser,24 and extracted case lists based on estrogen receptor (‘ER_Status_nature2012’), HER2 (‘HER2_Final_nature2012’), PAM50 (‘PAM50_mRNA_nature2012’), and histologic type (‘histological_type’). Cases with ‘Infiltrating Lobular Carcinoma’, ‘Mixed Histology’, ‘Mucinous Carcinoma’, ‘Other’, or missing histology were excluded. Somatic mutations of genes found to be mutated in neuroendocrine carcinomas were retrieved for estrogen receptor-positive (n=477), luminal A (n=183), luminal B (n=111), and estrogen receptor+/HER2− (n=376) from the cBioPortal website.25
For the METABRIC data set we retrieved the estrogen receptor and HER2 status, histology, and somatic mutations from the appropriate repository of Pereira et al.26, 27 PAM50 status was retrieved from the supplement of Curtis et al.28 Cases with ‘Mixed’, ‘Lobular’, ‘Other’, ‘NA’, ‘Mucinous’, ‘Benign’, or missing histology were excluded. Somatic mutations of genes found to be mutated in neuroendocrine carcinomas were extracted for estrogen receptor-positive (n=1282), luminal A (n=504), luminal B (n=378), and estrogen receptor+/HER2− (n=1125).
As the METABRIC data set provides information on histologic grade, we also compared the mutational frequencies of neuroendocrine carcinomas to those of estrogen receptor/HER2/grade-matched invasive carcinomas of no special type. METABRIC cases were estrogen receptor, HER2, and grade-matched to neuroendocrine carcinomas at a 1:5 ratio.
For all analyses, only mutations within the regions interrogated by the Ion AmpliSeq 22 cancer gene hotspot panel were considered. Comparisons were performed using Fisher’s exact tests. P-values<0.05 were considered statistically significant.
Prosigna Assay
For 35 cases, a 4-μm-thick section was prepared and stained with hematoxylin and eosin (H&E). A breast pathologist (JCT) identified the invasive tumor area and assessed tumor cellularity. Tumor cellularity was comprised between 20 and 80%. According to the manufacturer's instruction, depending on the surface area of the tumor a series of 10-μm sections were mounted onto Superfrost slides. RNA extraction and nCounter analysis were performed according to the Prosigna instructions.
Statistical Analysis
The R (version 3.2.1, cran, http://cran.r-project.org/) and R Studio software (version 7.6, http://www.rstudio.com, R Studio, Boston, USA) were used for the statistical analysis of immunohistochemical data. Statistical comparisons were performed using Student's t-test, Pearson correlation, and Fisher’s exact test. Survival curves are constructed with the Kaplan–Meier method. Progression-free survival was defined as the time between treatment and any recurrence or evidence of metastasis. Univariate survival analyses were done according to the Cox model with log-rank test. Two-sided tests with P<0.05 were considered statistically significant. The Cox proportional hazards regression model was used in the multivariate survival analysis that included lymph node status and therapy.
Results
Clinical Analysis
The clinical features of the 47 neuroendocrine carcinomas are described in Table 1. The mean age at diagnosis was 69 years, with a median of 67 years. Forty-one patients (87%) were postmenopausal. Seven patients (15%) had a history of ipsilateral or contralateral invasive carcinoma of no special type. Average tumor size was 23 mm (s.d.: 6–70), with a median of 20 mm. During the initial management, 43 patients (91%) underwent partial or total mastectomy as treatment. Three of the four patients who did not receive a first surgery had chemotherapy first; the other was treated with radiation and hormone therapy. Axillary dissection was performed in 39 patients (83%); there was a sentinel node biopsy for 14 of them, completed in six cases with axillary dissection. Radiotherapy was performed in 36 patients. Thirty-nine patients (83%) received hormonal therapy. Two patients received targeted therapy with Herceptin. One of these two patients showed a primary tumor with HER2 amplification confirmed by fluorescent in situ hybridization, the other showed a metastatic site harboring HER2 amplification.
Local recurrence was observed in one patient; metastatic disease was present in 12 patients, in three of whom it was presented at the time of initial diagnosis. The most frequent metastatic sites were bone, followed by lung, bone marrow, liver, pleura, skin, and two more unusual sites including the adrenal gland and pancreas.
Morphologic Features
The 47 neuroendocrine carcinomas were classified as follows: 30 well-differentiated carcinomas, seven poorly differentiated/small cell carcinomas, and 10 invasive breast carcinomas with neuroendocrine differentiation (two lobular carcinomas, five solid papillary carcinomas, three mucinous carcinomas of type B; Figure 1). Regarding the morphologic classification of our series: 79% were of no specific histologic type (other than showing neuroendocrine differentiation), 4% were lobular carcinomas, 11% were solid papillary carcinomas, and 6% were mucinous type B carcinomas (Figure 2). Twenty-nine (62%) were histologic grade 2, fifteen (32%) were grade 3, and three (6%) grade 1. The mean of mitotic count was 13 mitoses for 10 high-power fields, with a range of 1–111. Vascular invasion was observed in 25% of cases and three tumors showed perineural invasion. Necrosis was not observed in this series.

Examples of the three subtypes of neuroendocrine carcinomas. (a) Well-differentiated neuroendocrine carcinoma (H&E, × 40); (b) well-differentiated neuroendocrine carcinoma (H&E, × 400); (c) poorly differentiated neuroendocrine carcinoma (H&E, × 100); (d) poorly differentiated neuroendocrine carcinoma (H&E, × 200); (e) solid papillary carcinoma (H&E, × 100); (f) mucinous carcinoma (H&E, × 200).

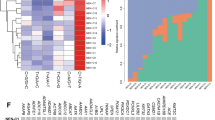
Overview of the cohort of neuroendocrine carcinomas. Heatmap illustrating the histologic and immunohistochemical features, IHC- and Prosigna-defined molecular subtype, sequencing results, cellularity of the samples, and clinical information. Cases are represented in columns; parameters are depicted in rows and color-coded according to the key. AR, androgen receptor; ER, estrogen receptor; NA, not available; NGS, next-generation sequencing; ROR, risk of recurrence; WT, wild type.
Six patients received neoadjuvant chemotherapy and the corresponding tumors were neuroendocrine carcinomas of no special type, where the tumor cellularity was high despite neoadjuvant chemotherapy. Cytotoxic effects were minimal in residual cells.
Immunophenotypic Analysis
Details on the immunophenotype of the 47 neuroendocrine carcinomas and comparison between different subtypes are described in detail in Table 2, Figure 2, and Supplementary Table 2.
Thirty-three tumors showed expression of chromogranin A (70%), 45 for synaptophysin (96%), and 31 both (66%; Figure 3). The mean expression of chromogranin A was 48% of the tumor cells with values ranging from 0 to 100% and a median of 50%. The mean synaptophysin expression was 84% of the tumor cells with values ranging from 1 to 100% and a median of 100%. All cases were estrogen receptor-positive (25–100% positive tumor cells) and the large majority (N=42, 89%) was also progesterone receptor-positive (1–100% positive tumor cells). One tumor (2%) was HER2-amplified and showed expression of both estrogen and progesterone receptors. The mean Ki67 proliferation index was 18% (range: 1–90%).

Immunohistochemistry for neuroendocrine carcinomas. (a) Chromogranin A (well-differentiated neuroendocrine carcinoma); (b) synaptophysin (poorly differentiated neuroendocrine carcinoma); (c) FOXA1 (mucinous carcinoma, strong nuclear stain); (d) GATA3 (poorly differentiated neuroendocrine carcinoma, strong nuclear stain).
When classified into molecular subtypes by immunohistochemistry, 24 cases (52%) were estrogen receptor+/HER2−/Ki67<14% (luminal A), 22 (48%) were estrogen receptor+/HER2−/Ki67>14% (luminal B-HER2 neg), and 1 was estrogen receptor+/HER2+/Ki67>14% (luminal B-HER2 pos).
The group of poorly differentiated neuroendocrine carcinomas was significantly more frequently of luminal B subtype (86%) compared with well-differentiated carcinomas (42%) (P=0.034).
All tumors except one (98%) expressed CK8/18. The large majority of tumors expressed GATA3 and FOXA1 (98% and 96%, respectively), similarly to the control group of invasive carcinomas of no special type (Figure 3 and Supplementary Table 3). None of the cases expressed basal markers (CK14, EGFR) nor TTF1.
All cases but one were CDX2-negative. The tumor that intensely expressed CDX2 was weakly estrogen receptor-positive and showed peripheral foci of ductal carcinoma in situ. Androgen receptor was significantly less frequently expressed in neuroendocrine carcinomas compared with the control group of invasive carcinomas of no special type (15% vs 100%, P<0.001, Supplementary Table 3) and whenever expressed, the intensity ranged from 1 to 3 and the percentage tumor cells showing an expression never exceeded 50%.
Clinical and immunohistochemical variables such as size, grade, nodal status, and Ki67 were not associated with a particular neuroendocrine subgroup or to GATA3 and FOXA1 expression.
Survival Analysis
Stratification of patients by histologic grade was statistically significant: the subset of grade 2 neuroendocrine carcinomas showed a better progression-free survival compared with that of grade 3 cases (HR=14.83, 95% CI 2.539–86.58, P=0.002; Figure 4a). Overall survival was not significantly different between these two groups. The survival analysis showed that patients with a neuroendocrine carcinoma had a progression-free survival significantly shorter than matched control series of patients with invasive carcinomas of no special type (HR=7.349, 95% CI 2.399–22.51, P=0.0005; Figure 4b). However there were no differences in terms of overall survival (P=0.52). Similar results for these two comparisons were obtained when these analyses were performed after excluding the patients with history of ipsilateral or contralateral invasive carcinoma of no special type as well as the patients affected by a carcinoma of histologic special type (Figure 5a and b).

Survival curves for the whole cohort (N=47) of neuroendocrine carcinomas. (a) PFS of neuroendocrine carcinomas stratified by grade; (b) PFS of neuroendocrine carcinomas matched to patients with invasive carcinomas of no special type; (c) overall survival of neuroendocrine carcinomas stratified by histologic subtype (30 well-differentiated neuroendocrine carcinomas, 7 poorly differentiated neuroendocrine carcinomas, and 10 carcinomas with neuroendocrine differentiation). NEBC, neuroendocrine breast carcinoma; NST BC, breast carcinoma of no special type; PFS, progression-free survival.

Survival analysis for a restricted cohort of neuroendocrine carcinomas. (a) After excluding the patients with a history of ipsilateral or contralateral invasive carcinoma of no special type as well as the patients affected by a carcinoma of histologic special type, the subset of grade 2 carcinomas still showed a better PFS compared with that of grade 3 cases (HR=12.39, 95% CI 1.66–92.18, P=0.01). (b) After excluding the patients with a history of ipsilateral or contralateral invasive carcinoma of no special type as well as the patients affected by a carcinoma of histologic special type, patients with a neuroendocrine carcinoma still had a PFS significantly shorter than matched control series of patients with invasive carcinomas of no special type (HR=8.37, 95% IC 1.72–40.99, P=0.008). (c) We did not reveal any differences in PFS (P=0.35) between well-differentiated, poorly differentiated neuroendocrine carcinomas, and invasive carcinomas with neuroendocrine differentiation after excluding patients with a history of ipsilateral or contralateral invasive carcinoma of no special type. NEBC, neuroendocrine breast carcinoma; NST BC, breast carcinoma of no special type; PFS, progression-free survival.
We did not observe any differences in overall survival (P=0.4) or progression-free survival (P=0.42) between well-differentiated, poorly differentiated neuroendocrine carcinomas, and invasive carcinomas with neuroendocrine differentiation (Figure 4c). Similar results were obtained when the analysis was run excluding the patients with a history of ipsilateral or contralateral invasive carcinoma of no special type (Figure 5c).
The multivariate survival analysis that included as variables lymph node status and therapy showed that neuroendocrine differentiation was an independent predictor of poor prognosis when we assessed progression-free survival (Supplementary Table 4). This analysis was performed on the cohort after exclusion of patients with a history of ipsilateral or contralateral invasive carcinoma of no special type as well as the patients affected by a carcinoma of histologic special type.
Molecular Analysis
Targeted sequencing using the Ion AmpliSeq 22 cancer gene hotspot panel
Targeted sequencing analysis was performed at a mean depth of 2256 × (Supplementary Table 5) and 85% of analyzed cases showed a depth above 500 ×.
PIK3CA mutations were observed in three cases (3/42, 7%) with classical somatic activating hotspot mutations described in the COSMIC repository, of which two were in exon 20 (kinase domain), namely a H1047R and a H1047L. The third mutation was located in exon 9 in the helical domain of the catalytic subunit p110α, i.e., a E542K. The detected allele frequencies were 10%, 40% and 5%, respectively. The three PIK3CA mutations were all found in well-differentiated neuroendocrine tumors.
Three other cases (3/42, 7%) harbored a TP53 mutation (C277Y, Y220C, and H193R) with an allele frequency of 5%, 30%, and 9%, respectively. These three TP53 mutations are described in the COSMIC repository. Two out of the three TP53-mutated cases were poorly differentiated neuroendocrine carcinomas (Figure 2).
All of other tested genes were not mutated in the current cohort, in the limit of the analyzed sequences.
We next compared the PIK3CA and TP53 mutational frequencies found in neuroendocrine carcinomas with those of invasive breast carcinomas from TCGA and METABRIC, considering only the mutations within the regions interrogated by the Ion AmpliSeq 22 cancer gene hotspot panel. PIK3CA mutations were significantly less frequent in neuroendocrine carcinomas compared with luminal A, luminal B, and estrogen receptor-positive carcinomas from both TCGA and METABRIC, as well as from a cohort of invasive carcinomas of no special type matched for estrogen receptor/HER2 status and histologic grade extracted from the METABRIC data set (all P-values <0.05, Fisher’s exact tests, Table 3).
TP53 mutations were significantly less prevalent in neuroendocrine carcinomas compared with luminal B from both TCGA and METABRIC (P=0.006 and 0.001, respectively, Fisher’s exact test), as well as from estrogen receptor-positive carcinomas from TCGA (P=0.01, Fisher’s exact test) and from the cohort of invasive carcinomas of no special type matched for estrogen receptor/HER2 status and histologic grade (P=0.048, Fisher’s exact test).
Prosigna analysis and assessment of risk of recurrence
For the 35 neuroendocrine carcinomas tested with Prosigna, the distribution of molecular classes resulted in 18 luminal A tumors (51%), 16 luminal B tumors (46%), and one basal-like tumor (3%). The CDX2-positive carcinoma was classified as a luminal A carcinoma. The concordance rate of molecular and immunohistochemical classification was 86%.
The five cases (14%) differently classified by Prosigna consisted of two IHC-luminal A carcinomas re-classified as luminal B; three IHC-luminal B re-classified as two luminal A, and one basal-like (Figure 2). The latter showed estrogen receptor expression in about 25% of tumor cells and was a poorly differentiated neuroendocrine carcinoma, whereas the others were all well-differentiated neuroendocrine carcinomas. One of the two cases re-classified as luminal A by Prosigna had a tumor cellularity of 30% (Figure 2).
In addition, 11 tumors (31%) were classified as low risk with a mean risk of recurrence (ROR) at 10 years of 4%; all of them were luminal A tumors (Figure 2). Seven tumors (20%) were classified as intermediate risk with a mean ROR at 10 years of 8%. Finally, 17 tumors (49%) were classified as high risk with a mean 10-year ROR of 31%. In the Prosigna high risk group, 14 tumors were luminal B and three luminal A molecular subtype (Figure 2). A percentage of tumors (18%) classified as luminal A (either by immunohistochemistry or by Prosigna) had a high ROR.
We finally compared the ROR distribution in relation to the events observed in our cohort and observed no significant difference of ROR distribution between patients with or without disease progression or death of disease (Figure 6). This result has to be taken with caution as only one patient experienced a local relapse and 12 had distant metastasis; in addition, the follow-up of the patients was shorter than 10 years.

Risk of recurrence (ROR) distribution and patient survival. There were no significant differences in ROR distribution between patients with or without disease progression or death of disease.
Discussion
In this study we confirm that neuroendocrine breast carcinomas can belong to either the luminal A or luminal B subgroup; however, in stark contrast to common forms of luminal carcinomas, they harbor a significantly lower frequency of PIK3CA mutation. In addition, our clinical data highlight a worse progression-free survival of patients affected by neuroendocrine carcinomas as compared with that of patients with an invasive breast carcinoma of no special type matched for age, size, grade, and estrogen receptor status.
A strong association between neuroendocrine breast carcinomas and expression of hormone receptors has been repeatedly shown by previous immunohistochemical studies.29, 30, 31 At the transcriptomic level, Weigelt et al have described neuroendocrine carcinomas to form a separate cluster together with mucinous type A and mucinous type B tumors within the luminal subgroups.16
Here we found an almost equal distribution of luminal A and B (52% and 48%, respectively) tumors by Prosigna classification. Few studies have investigated the luminal A/B distinction within neuroendocrine carcinomas. In a limited cohort of six samples, by gene expression profiling analysis Weigelt et al have reported neuroendocrine carcinomas to pertain to the luminal A more frequently than to the luminal B subtype.32 In a larger series, Bogina et al13 have recently reported that 58% of the analyzed neuroendocrine carcinomas were of luminal B-like subtype, with the molecular classes being determined by immunohistochemistry as a surrogate for gene expression.
As all tumors but one were luminal, we next gained insight into complete luminal differentiation of these tumors. We showed that neuroendocrine carcinomas are usually GATA3-, FOXA1-, and CK8/18-positive. FOXA1 and GATA3 expression was comparable between neuroendocrine carcinomas and the matched cohort of invasive carcinomas of no special type; this is not surprising as GATA3 and FOXA1 are usually expressed in luminal breast carcinomas.33 As GATA3 and FOXA1 expression is lower in poorly differentiated and high-grade cancers,34, 35 we investigated possible differences among the categories of neuroendocrine carcinomas: there were no significant associations between the differentiation of the neuroendocrine tumor, histologic grade, and FOXA1 or GATA3 expression.
We also report on the expression of androgen receptor in neuroendocrine carcinomas, which was found in 15% of our series, and this was significantly lower than that in invasive carcinomas of no special type (100%). Our results are in line with a previous study on neuroendocrine carcinomas demonstrating androgen receptor expression in 18% of cases.8
Taken together, the results of this immunophenotypic characterization show that breast tumors can develop simultaneously to several differentiation pathways both endocrine and exocrine. A mixed differentiation with neuroendocrine and exocrine protein production is a phenomenon quite common and known in the tumors of endocrine and non-endocrine organs.36, 37, 38
The landscape of genetic alterations of neuroendocrine carcinomas of the breast is yet to be fully determined as molecular studies are scarce. At present, one study has focused on point mutations of a small series of neuroendocrine carcinomas and showed recurrent mutations affecting PIK3CA and the FGFR family (FGFR1 and FGFR4).17 HRAS and KDR mutations were observed in single cases.17 A subsequent analysis has recently showed that neuroendocrine carcinomas seem to harbor a repertoire of somatic mutations distinct from that of common types of estrogen receptor-positive/HER2-negative breast carcinomas, featuring, for instance, lower frequencies of TP53 and PIK3CA mutations18 and a higher frequency of mutations in chromatin-remodeling genes, such as ARID1A. Regrettably, with the Ion AmpliSeq 22 cancer gene hotspot panel used in the present study, we could not investigate the prevalence of mutations in chromatin-remodeling genes in our cohort; however, we were able to explore the frequency of both TP53 and PIK3CA mutations. It is important to note that all of the comparative analyses were performed by considering only the mutations within the regions interrogated in the Ion AmpliSeq 22 cancer gene hotspot panel used in the present study. Our targeted sequencing analysis identified a rate of TP53 mutations (7%, three cases) significantly lower than that of luminal B carcinomas from TCGA (31%) and METABRIC (26%) as well as of a matched series of estrogen receptor-positive/HER2-negative invasive carcinomas of no special type from the METABRIC data set (20%). Interestingly, two of the three cases were poorly differentiated carcinomas, thus mirroring what has been observed in neuroendocrine carcinomas of other sites, such as for instance the lung, where TP53 is encountered in small cell lung carcinomas and seems to be absent or rare in lung carcinoids.39, 40
We also detected PIK3CA mutations in 7% of cases (all well-differentiated neuroendocrine carcinomas), a rate that is significantly lower than that of estrogen receptor-positive, luminal A, and luminal B carcinomas calculated from TCGA23 and METABRIC41 data sets. Furthermore, the prevalence of PIK3CA mutations in neuroendocrine carcinomas was found to be significantly lower than a series of invasive carcinomas of no special type matched for estrogen receptor, HER2 status, and histologic grade extracted from METABRIC (38%). As stressed above, it is crucial to highlight that when we compared the frequency of PIK3CA mutations we restricted the analysis to the regions assessed by the Ion AmpliSeq 22 cancer gene hotspot panel. Still, one could argue that we cannot exclude the possibility of false-negative results in our sequencing analysis due to a cellularity as low as 30% for five samples. Nevertheless, Marchiò et al18 have recently reported similar findings in a series of microdissected neuroendocrine carcinomas. Taken together, these independent observations support the concept that neuroendocrine carcinomas may represent a particular entity within the luminal cancer spectrum, similar to mucinous carcinomas of the breast, for which a low frequency of PIK3CA mutations has been reported in other studies.42 Interestingly, neuroendocrine carcinomas of the breast have been shown to share similar transcriptomic profiles with type B mucinous carcinomas.32 Altogether, these observations may suggest a putative common initiating cancer cell for these two entities or common etiologies.
Of note, several studies in the literature show a significant association between the presence of PIK3CA mutation and favorable clinicopathological characteristics such as small tumor size or low grade.43, 44 As a matter of fact, our clinical data support a worse prognosis in patients affected by neuroendocrine carcinomas as compared with that of patients with invasive breast carcinomas of no special type matched for age, size, grade, and estrogen receptor status. This was confirmed also in multivariate progression-free survival analysis taking into account lymph node status and therapy. A large proportion of our cohort (49%) showed a high ROR by Prosigna, with a mean 10-year ROR of 31%. Even in the luminal A subgroup, 18% of cases had a high ROR.
We finally investigated the impact of histologic subtyping of neuroendocrine breast carcinomas according to the 2012 WHO classification on long-term outcomes. Some have showed a worse prognosis of the poorly differentiated subgroup.45, 46 In our series, although overall survival and progression-free survival of poorly differentiated neuroendocrine carcinomas were worse compared with the other two groups, these differences were not statistically significant. However, we were limited by the size of our cohort (respectively, seven and ten cases of poorly differentiated neuroendocrine carcinoma and carcinoma with neuroendocrine differentiation). Therefore, larger studies are warranted to ascertain a possible role of such classification.
In conclusion, we have shown that the rare variant of neuroendocrine breast cancer occurs predominantly in postmenopausal women and is associated with aggressive locoregional and metastatic evolution, including unusual sites (adrenal gland), with a poorer progression-free survival than invasive carcinomas of no special type. At the molecular level, the subgroup of neuroendocrine carcinomas is part of the spectrum of luminal carcinomas and is characterized by low levels of PIK3CA mutations. This molecular feature may contribute to partly explain the unfavorable prognosis of neuroendocrine breast carcinomas. Larger molecular-profiling studies of this entity with correlation with clinical data are warranted.
Currently, the clinical decision making for patients affected by a neuroendocrine carcinoma is based on proper grading and immunophenotyping of the lesions, similar to any invasive carcinomas of no special type. Nevertheless, our results also suggest that an accurate identification of this entity may be useful to better tailor patient adjuvant therapy within luminal carcinomas.
References
Feyrter F, Hartmann G . [on the carcinoid growth form of the carcinoma mammae, especially the carcinoma solidum (Gelatinosum) mammae]. Frankf Z Pathol 1963; 73: 24–39.
Tang F, Wei B, Tian Z et al, Invasive mammary carcinoma with neuroendocrine differentiation: histological features and diagnostic challenges. Histopathology 2011; 59: 106–115.
Woodard BH, Eisenbarth G, Wallace NR et al, Adrenocorticotropin production by a mammary carcinoma. Cancer 1981; 47: 1823–1827.
Ellis IO, Schnitt SJ, Sastre Garau X et al, Neuroendocrine tumours. In: Tavassoli F, Devilee P (eds). Pathology and Genetics of Tumours of the Breast and Female Genital Organs. IARC Press: Lyon, France, 2003, pp 32–34.
Bussolati G, Badve S. Carcinoma with neuroendocrine features. In: Lakhani S, Ellis IO, Schnitt SJ et al, (eds). WHO Classification of Tumours of the Breast. IARC Press: Lyon, France, 2012, pp 62–63.
Maluf HM, Koerner FC . Carcinomas of the breast with endocrine differentiation: a review. Virchows Arch 1994; 425: 449–457.
Maluf HM, Koerner FC . Solid papillary carcinoma of the breast. A form of intraductal carcinoma with endocrine differentiation frequently associated with mucinous carcinoma. Am J Surg Pathol 1995; 19: 1237–1244.
Sapino A, Righi L, Cassoni P et al, Expression of apocrine differentiation markers in neuroendocrine breast carcinomas of aged women. Mod Pathol 2001; 14: 768–776.
Makretsov N, Gilks CB, Coldman AJ et al, Tissue microarray analysis of neuroendocrine differentiation and its prognostic significance in breast cancer. Hum Pathol 2003; 34: 1001–1008.
Miremadi A, Pinder SE, Lee AH et al, Neuroendocrine differentiation and prognosis in breast adenocarcinoma. Histopathology 2002; 40: 215–222.
van Krimpen C, Elferink A, Broodman CA et al, The prognostic influence of neuroendocrine differentiation in breast cancer: results of a long-term follow-up study. Breast 2004; 13: 329–333.
Wang J, Wei B, Albarracin CT et al, Invasive neuroendocrine carcinoma of the breast: a population-based study from the surveillance, epidemiology and end results (SEER) database. BMC Cancer 2014; 14: 147.
Bogina G, Munari E, Brunelli M et al, Neuroendocrine differentiation in breast carcinoma: clinicopathological features and outcome. Histopathology 2016; 68: 422–432.
Rovera F, Lavazza M, La Rosa S et al, Neuroendocrine breast cancer: retrospective analysis of 96 patients and review of literature. Int J Surg 2013; 11 (Suppl 1): S79–S83.
Rovera F, Masciocchi P, Coglitore A et al, Neuroendocrine carcinomas of the breast. Int J Surg 2008; 6 (Suppl 1): S113–S115.
Weigelt B, Horlings HM, Kreike B et al, Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol 2008; 216: 141–150.
Ang D, Ballard M, Beadling C et al, Novel mutations in neuroendocrine carcinoma of the breast: possible therapeutic targets. Diagn Mol Pathol 2014; 23: 97–103.
Marchio C, Geyer FC, Ng CK et al, The genetic landscape of breast carcinomas with neuroendocrine differentiation. J Pathol 2017; 241: 405–419.
Elston CW, Ellis IO . Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 1991; 19: 403–410.
Hammond ME, Hayes DF, Dowsett M et al, American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 2010; 28: 2784–2795.
Wolff AC, Hammond ME, Hicks DG et al, Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013; 31: 3997–4013.
Goldhirsch A, Wood WC, Coates AS et al, Strategies for subtypes—dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol 2011; 22: 1736–1747.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70.
UCSC Xena Browserhttps://xenabrowser.net/datapages/?cohort=TCGABreast Cancer (BRCA).(Accessed 5 April 2017).
cbioportal.http://www.cbioportal.org(Accessed 19 April 2017).
https://github.com/cclab-brca/mutationalProfiles(Accessed 1 November 2016).
Pereira B, Chin SF, Rueda OM et al, The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016; 7: 11479.
Curtis C, Shah SP, Chin SF et al, The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486: 346–352.
Kwon SY, Bae YK, Gu MJ et al, Neuroendocrine differentiation correlates with hormone receptor expression and decreased survival in patients with invasive breast carcinoma. Histopathology 2014; 64: 647–659.
Righi L, Sapino A, Marchio C et al, Neuroendocrine differentiation in breast cancer: established facts and unresolved problems. Semin Diagn Pathol 2010; 27: 69–76.
Zhang Y, Chen Z, Bao Y et al, Invasive neuroendocrine carcinoma of the breast: a prognostic research of 107 Chinese patients. Neoplasma 2013; 60: 215–222.
Weigelt B, Geyer FC, Horlings HM et al, Mucinous and neuroendocrine breast carcinomas are transcriptionally distinct from invasive ductal carcinomas of no special type. Mod Pathol 2009; 22: 1401–1414.
Asselin-Labat ML, Sutherland KD, Barker H et al, Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol 2007; 9: 201–209.
Hisamatsu Y, Tokunaga E, Yamashita N et al, Impact of FOXA1 expression on the prognosis of patients with hormone receptor-positive breast cancer. Ann Surg Oncol 2012; 19: 1145–1152.
Yoon YS, Kim SY, Lee JH et al, Primary neuroendocrine carcinoma of the breast: radiologic and pathologic correlation. Clin Imaging 2014; 38: 734–738.
Brambilla E, Lantuejoul S, Sturm N . Divergent differentiation in neuroendocrine lung tumors. Semin Diagn Pathol 2000; 17: 138–148.
Capella C, Usellini L, Papotti M et al, Ultrastructural features of neuroendocrine differentiated carcinomas of the breast. Ultrastruct Pathol 1990; 14: 321–334.
di Sant' Agnese PA . Divergent neuroendocrine differentiation in prostatic carcinoma. Semin Diagn Pathol 2000; 17: 149–161.
Rekhtman N, Pietanza MC, Hellmann MD et al, Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin Cancer Res 2016; 22: 3618–3629.
Simbolo M, Mafficini A, Sikora KO et al, Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J Pathol 2017; 241: 488–500.
Pereira B, Chin SF, Rueda OM et al, Erratum: the somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat Commun 2016; 7: 11908.
Kehr EL, Jorns JM, Ang D et al, Mucinous breast carcinomas lack PIK3CA and AKT1 mutations. Hum Pathol 2012; 43: 2207–2212.
Cizkova M, Susini A, Vacher S et al, PIK3CA mutation impact on survival in breast cancer patients and in ERalpha, PR and ERBB2-based subgroups. Breast Cancer Res 2012; 14: R28.
Kalinsky K, Jacks LM, Heguy A et al, PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res 2009; 15: 5049–5059.
Cloyd JM, Yang RL, Allison KH et al, Impact of histological subtype on long-term outcomes of neuroendocrine carcinoma of the breast. Breast Cancer Res Treat 2014; 148: 637–644.
Shin SJ, DeLellis RA, Ying L et al, Small cell carcinoma of the breast: a clinicopathologic and immunohistochemical study of nine patients. Am J Surg Pathol 2000; 24: 1231–1238.
Acknowledgements
CM is supported by the Italian Ministry of University (ex-60% 2014 and 2015). We thank Eliane Padoy and Myriam Chaillou for their help in retrieving the tumor blocks.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Lavigne, M., Menet, E., Tille, JC. et al. Comprehensive clinical and molecular analyses of neuroendocrine carcinomas of the breast. Mod Pathol 31, 68–82 (2018). https://doi.org/10.1038/modpathol.2017.107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.107
This article is cited by
-
Clinical outcome and therapeutic impact on neuroendocrine neoplasms of the breast: a national cancer database study
Breast Cancer Research and Treatment (2023)
-
Clinical and genomic analyses of neuroendocrine neoplasms of the breast
Modern Pathology (2022)
-
Comparison of the mutational profiles of neuroendocrine breast tumours, invasive ductal carcinomas and pancreatic neuroendocrine carcinomas
Oncogenesis (2022)
-
Genetic and immunohistochemical profiling of small cell and large cell neuroendocrine carcinomas of the breast
Modern Pathology (2022)
-
Comprehensive analysis of clinicopathologic features and p53 mutation in neuroendocrine neoplasms of the breast: experience from a large academic center
Breast Cancer Research and Treatment (2022)
