
Abstract
In 2010, the World Health Organization reclassified the entity originally described as intraductal oncocytic papillary neoplasm as the ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm. Although several key molecular alterations of other intraductal papillary mucinous neoplasm subtypes have been discovered, including common mutations in KRAS, GNAS, and RNF3, those of oncocytic subtype have not been well characterized. We analyzed 11 pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms. Nine pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms uniformly exhibited typical entity-defining morphology of arborizing papillae lined by layers of cells with oncocytic cytoplasm, prominent, nucleoli, and intraepithelial lumina. The remaining two were atypical. One lacked the arborizing papilla and had flat oncocytic epithelium only; the other one had focal oncocytic epithelium in a background of predominantly intestinal subtype intraductal papillary mucinous neoplasm. Different components of this case were analyzed separately. Formalin-fixed, paraffin-embedded specimens of all cases were microdissected and subjected to high-depth-targeted next-generation sequencing for a panel of 300 key cancer-associated genes in a platform that enabled the identification of sequence mutations, copy number alterations, and select structural rearrangements involving all targeted genes. Fresh frozen specimens of two cases were also subjected to whole-genome sequencing. For the nine typical pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms, the number of mutations per case, identified by next-generation sequencing, ranged from 1 to 10 (median=4). None of these cases had KRAS or GNAS mutations and only one had both RNF43 and PIK3R1 mutations. ARHGAP26, ASXL1, EPHA8, and ERBB4 genes were somatically altered in more than one of these typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms but not in the other two atypical ones. In the neoplasm with flat oncocytic epithelium, the only mutated gene was KRAS. All components of the intestinal subtype intraductal papillary mucinous neoplasms with focal oncocytic epithelium manifested TP53, GNAS, and RNF43 mutations. In conclusion, this study elucidates that ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm is not only morphologically distinct but also genetically distinct from other intraductal papillary mucinous neoplasm subtypes. Considering that now its biologic behavior is also being found to be different than other intraductal papillary mucinous neoplasm subtypes, ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm warrants being recognized separately.
Similar content being viewed by others
Main
‘Oncocytic subtype’ of intraductal papillary mucinous neoplasms of the pancreas was originally described as a separate variant of pancreatic intraductal neoplasms.1 However, the current (2010) World Health Organization designated this neoplasm as a subtype of intraductal papillary mucinous neoplasm of the pancreas because oncocytic subtype has some overlapping features with other subtypes of intraductal papillary mucinous neoplasm.2 For example, they all present as an at least partially cystic pancreatic mass.3 The cystic appearance is due to dilation of the ducts by an intraductal neoplasm composed of tumor cells arranged in a papillary pattern.1, 4 These neoplasms also have less aggressive course than conventional pancreatic ductal adenocarcinoma, even if there is an associated invasive carcinoma.1, 3, 4, 5, 6, 7, 8, 9, 10 In fact, the family of intraductal papillary mucinous neoplasms may contain ‘individuals’, which show overlapping features between the four so far recognized subtypes.
However, ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm also has several distinguishing pathologic characteristics such as the complex arborizing papillae with delicate fibrovascular cores and distinctive intraepithelial lumina formation, in addition to the oncocytic nature of the cells, due to abundant intracytoplasmic mitochondria.1, 3, 4, 9, 11 In other organs, tumors that are prone to accumulate abundant intracytoplasmic mitochondria in the exclusion of other organelles appear to have distinct pathogenesis and different biology than their non-oncocytic counterparts, with the best example being renal oncocytic neoplasms or Hurthle cell tumors of the thyroid.12, 13 Recently, the studies have shown that despite being very complex lesions, ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms are also actually either non-invasive or minimally invasive, and although they may recur, their seldom lead to mortality of the patient.1, 9, 10
With the introduction of routine molecular genetic analyses, including next-generation sequencing,14, 15, 16 various molecular alterations have been identified in other subtypes of intraductal papillary mucinous neoplasms. As in many ductal neoplasms including pancreatic ductal adenocarcinomas for which KRAS mutation appears to be a nearly pre-requisite baseline change, KRAS mutations are the most common mutations and have been detected in the majority of intraductal papillary mucinous neoplasms (up to 100%).8, 15, 17, 18, 19, 20, 21, 22 More interestingly, activating GNAS mutations at codon 201 have been identified in approximately half (41–66%) of intraductal papillary mucinous neoplasms,15, 16, 23, 24, 25, 26 particularly in the intestinal subtype.15, 23, 25 Inactivating mutations in the RNF43 gene, a likely tumor suppressor and negative regulator of the Wnt signaling pathway,27 are also seen in up to 75% of intraductal papillary mucinous neoplasms.15, 24 Mutations of PIK3CA are described in 3–11%.28, 29, 30 Less common alterations involve CDKN2A/p16 (loss of expression in 18% of non-invasive intraductal papillary mucinous neoplasms and 53% of associated invasive carcinomas), SMAD4 (loss of expression in 3% of non-invasive intraductal papillary mucinous neoplasms and 30% of associated invasive carcinomas), TP53 (10% of high-grade intraductal papillary mucinous neoplasms), BRAF (6% of high-grade intraductal papillary mucinous neoplasms), CTNNB1/β-catenin (4%), IDH1 (4%), STK11 (4%), PTEN (4%), ATM (2%), CDH1 (2%), FGFR3 (2%), and SRC (2%).17, 24
In contrast, the literature on the molecular features of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm is fairly limited, partially due to relative rarity of the neoplasm. Emerging studies including case reports,31 a few cases studied together with other subtypes of intraductal papillary mucinous neoplasms,15, 32, 33 or studies analyzing selected gene mutations, such as KRAS,34, 35 have shown that ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm generally lacks the mutations commonly found in other subtypes of intraductal papillary mucinous neoplasms. This raises the questions of whether ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm is genetically distinct from the other subtypes of intraductal papillary mucinous neoplasm and furthermore, whether it should be considered as a separate diagnostic entity?36 To further elucidate this question, we analyzed a series of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms by targeted next-generation sequencing, using a broad panel of cancer-related genes, to investigate genes not previously assessed.
Materials and Methods
With approval of the Institutional Review Board, the surgical pathology databases of Memorial Sloan Kettering Cancer Center and Emory University were searched for patients with a diagnosis of pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm between 1991 and 2013. Resections of 11 pancreatic neoplasms were identified for which the slides and tissue blocks were available. The diagnoses were confirmed by the authors. Medical records including pathology reports were reviewed to obtain clinical data including age, gender, treatment modalities, and outcome.
Targeted Next-Generation Sequencing
Twenty 10-micron-thick sections were cut from formalin-fixed, paraffin-embedded tissue blocks containing ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms. From these sections, areas of interest were needle microdissected. For each patient, extraction of DNA was performed on dissected tissue and where available on normal, non-pancreatic tissue (stomach, spleen or duodenum). Deep coverage, targeted next-generation sequencing was then performed on a panel of 300 genes, including KRAS, GNAS, and RNF43, listed in the supporting information (Supplementary Information File 1), known to undergo somatic genomic alterations in cancer, as previously described.37, 38 Briefly, massively parallel sequencing libraries (Kapa Biosystems, New England Biolabs) that contain barcoded universal primers were generated from 115 to 250 ng genomic DNA from the tumor material and matched normal tissue. After library amplification and DNA quantification, equimolar pools were generated consisting of up to 24 barcoded libraries. These DNA pools were subjected to solution-phase hybrid capture with synthetic biotinylated DNA probes (Nimblegen SeqCap) targeting all protein-coding exons from the selected 300 cancer genes as well as introns known to harbor recurrent translocation breakpoints. Genes were selected to include commonly implicated oncogenes, tumor suppressor genes, and members of pathways deemed actionable by targeted therapies. Each hybrid capture pool was sequenced to deep coverage in a single paired-end lane of an Illumina flow cell. Subsequently, the sequencing data were deconvoluted to match all high-quality barcoded reads with the corresponding tumor samples, and genomic alterations (single-nucleotide sequence variants, small insertions/deletions, and DNA copy number alterations) were identified. For matched tumor/normal tissue pairs (n=9), somatic single-nucleotide variants and insertions and deletions were called using MuTect and the SomaticIndelDetector tools in GATK, respectively.39, 40 For unmatched tumors (n=2), MuTect was run against a pool of unrelated DNAs from normal formalin-fixed, paraffin-embedded tissue blocks, and variants were filtered out if they were present in the 1000 Genomes project at a population frequency of >1%. All candidate mutations, insertions, and deletions were reviewed manually using the Integrative Genomics Viewer.41
Whole-Genome Sequencing
Fresh frozen tumor material and matched normal tissues of two cases (Cases 4 and 5) were also subjected to whole-genome sequencing, which was performed using Illumina paired-end chemistry on a HiSeqX sequencer, which yielded coverage of at least 80 × for tumor samples and 40 × for normal samples, with more than 95% of the target bases having at least 5 × coverage.
Genomic DNA isolation
Tissue was extracted using Qiagen AllPrep DNA/RNA Mini Kit. DNA was quantified using the Qubit 2.0 Fluorometer, Invitrogen, and quality was determined by using Agilent Bioanlyzer.
Illumina whole-genome sequencing and genotyping
DNA libraries were prepared using the KAPA Hyper Prep Kit (Kapa, Kapa Biosystems, Wilmington, MA, USA). For each sample library preparation, 100 ng of high molecular weight genomic DNA was fragmented using the Covaris LE220 system to an average size of 350 bp. Fragmented samples were end repaired and adenylated using Kapa’s end-repair and a-tailing enzymes. The samples were then ligated with Biooscientific adapters and PCR amplified using KAPA Hifi HotStart Master Mix (Kapa, Kapa Biosystems, Wilmington, MA, USA). The DNA libraries were clustered onto flowcells using Illumina’s cBot and HiSeq Paired End Cluster Generation kits as per manufacturer protocol (Illumina, San Diego, CA, USA). Sequencing was performed using 2 × 150 Illumina HiSeqX platform with v2.5 chemistry reagents. Genotyping was performed using HumanOmni2.5 M BeadChips (Illumina).
Whole-genome sequencing and data analysis was performed at the New York Genome Center (NYGC, New York, NY, USA). Paired-end 2 × 150 bp reads were aligned to the GRCh37 human reference using the Burrows-Wheeler Aligner (BWA aln v.0.7.8)42 and processed using the best-practices pipeline that includes marking of duplicate reads by the use of Picard tools and realignment around indels and base recalibration via Genome Analysis Toolkit (GATK) ver. 2.7.4.43 We employ the following variant callers: muTect v1.1.4,39 LoFreq v2.0.044 (single-nucleotide variants only), Strelka v1.0.1345 (both single-nucleotide variants and indels), Pindel46 and Scalpel47 (indels only) and return the union of calls, filtered using the default filtering criteria as implemented in each of the callers. Single-nucleotide variants and indels were annotated via snpEff, snpSift,48 and GATK VariantAnnotator using annotation from ENSEMBL, COSMIC,49 Gene Ontology, and 1000 Genomes.
Structural variants, such as copy number variants as well as complex genomic rearrangements, were detected by the use of multiple tools: NBIC-seq50 for copy number variants/structural variants calling, Delly,51 Crest,52 and BreakDancer53 for structural variant calling. We prioritize structural variants in the intersection of callers and structural variants for which we can find additional split-read evidence using SplazerS.54 Structural variants for which there is split-read support in the matched normal or that are annotated as known germline variants (1000 Genomes call set, DGV) were removed as likely remaining germline variants. The predicted sets of somatic structural variants were annotated with gene overlap (RefSeq, Cancer Gene Census) including prediction of potential effect on genes (eg, disruptive/exonic, intronic, intergenic, fusion candidate). In addition, copy number variants and loss of heterozygosity were also analyzed from the genotyping chip using Nexus (Biodiscovery) software.
Results
Clinicopathologic Features
Eleven patients were identified. The clinicopathologic features of the cases are summarized in Table 1. Half of these patients were male and the mean age was 60 years (range, 45–78). No patients received neoadjuvant chemotherapy or chemoradiation. One patient with an associated invasive colloid carcinoma received adjuvant chemotherapy, but not chemoradiation. The majority (67%) of tumors was located in the head and patients underwent pancreatoduodenectomy. Tumor size varied from 1 to 10 cm (median, 5.5 cm).
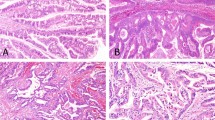
Of 11 cases, 9 exhibited typical entity-defining characteristics. Grossly, these tumors were characterized by large, soft tan friable nodules associated with cystic spaces (Figure 1). In contrast to other subtypes of intraductal papillary mucinous neoplasm, the intraductal location was difficult to recognize. Microscopically, the tumors were architecturally complex, with arborizing papillae growing into the lumens of massively dilated ducts. The neoplastic epithelial cells had abundant eosinophilic granular cytoplasm and nuclei with single prominent nucleoli. Intraepithelial and intracellular lumina were also identified (Figure 2). Only one of the nine typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm had an associated invasive carcinoma, in the form of multiple microscopic foci of small micropapillary clusters of oncocytic cells infiltrating the periductal stroma. The invasive component was too small for molecular analysis.

Macroscopically, typical pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms were characterized by tan, friable, and large papillary excrescences filling cystic structures.

Papillae of typical pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms were often very delicate and arborizing (a), and the cells revealed distinctive oncocytic appearance with voluminous acidophilic granular cytoplasm and single prominent nucleoli. (b) Multiple intracellular lumina were also present.
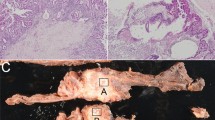
Two ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms had atypical morphology. Case 10 (Table 2) was non-papillary and revealed a flat cyst-lining of neoplastic epithelial cells with oncocytic features (Figure 3). Case 11 (Table 2) was predominantly intestinal subtype intraductal papillary mucinous neoplasm with a distinct focus of oncocytic epithelium on H&E (Figure 4). Of note, a previously performed MUC6 immunohistochemical stain was negative in the intestinal component and positive only at the base of the papillae of the oncocytic focus. This case also had an associated invasive colloid carcinoma (Figure 5). All components of this case (intestinal subtype intraductal papillary mucinous neoplasm, oncocytic, and invasive carcinoma) were analyzed separately.

Case 10 lacked the arborizing papillae (a) but the neoplastic epithelial cells revealed oncocytic features (b).

Case 11 was a predominantly intestinal subtype intraductal papillary mucinous neoplasm (a) with a distinct focus of oncocytic epithelium (b). Intestinal and oncocytic components were analyzed separately.

Case 11 also revealed an associated invasive carcinoma of colloid type characterized by pools of mucin that are partially lined by carcinoma cells.
The follow-up period ranged from 2 to 190 months (median, 61 months). Two of nine patients with available follow-up died; the remaining seven (77%) were alive without disease. Case 11 (with heteregenous epithelium and an associated invasive colloid carcinoma) died of perioperative complications 2 months after the surgery, and Case 2 (typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm) died of unrelated causes after 190 months.
Molecular Features
Targeted next-generation sequencing
A total of 13 tumor samples from 11 patients underwent targeted deep sequencing. The results of these genetic studies are summarized in Table 2, and details of the individual cases are described in the supporting information (Supplementary Information File 2).
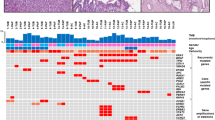
A total of 41 mutations were identified in the 11 cases, ranging 1–10 mutations per neoplasm (median=4, Figure 6). In the nine typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms, the following four genes were mutated in at least two neoplasms: ARHGAP26 (missense mutations at K592N or P4Q), ASXL1 (missense mutation at V119L or a frameshift deletion at M341fs), EPHA8 (missense mutations at R375H or R384H), and ERBB4 (missense mutations at L939F or L1163M). None of the nine typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms had KRAS or GNAS mutations. One typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm (Case 2) had RNF43 (frameshift/deletion at L61fs), PIK3R1 (a missense mutation at P194T), and PIK3R3 (missense mutation at R49Q) mutations.

Number of mutations seen in each case analyzed.
In case 10 (with flat oncocytic epithelium), the only somatic mutation found was KRAS (missense mutation at G12V). Case 11 (with heteregenous epithelium and an associated invasive colloid carcinoma) manifested GNAS (missense mutation at R201C and R201S), RNF43 (missense mutation at M1I), and TP53 (missense mutations at R248W and S241F as well as nonsense mutation at W146) mutations in all three components. Whereas, PDGFRA (missense mutation at T230M) and ATRX (missense mutation at E1492G) mutations were exclusive to the intestinal component.
Whole-genome sequencing
Fresh frozen tumor samples from two patients (Cases 4 and 5) also underwent whole-genome sequencing. Details of the individual cases are described in the supporting information (Supplementary Information Files 3 and 4).
Copy number analysis revealed that both samples had multiple copy number gains and losses, the vast majority of which including a contiguous set of genes, in multiple chromosomes (Case 4: chromosomes 8, 12, 17, and 21; Case 5: chromosomes 1, 2, 3, 7, 8, 9, 13, 14, 15, and 22). Also both specimens had clonal loss of heterozygosity of an entire chromosome (Case 4: chromosome 12; Case 5: chromosome 20).
A total of 91 mutations within 87 genes were identified among the 2 cases, 40 mutations in Case 4 and 51 mutations in Case 5. Among the five gene mutations in Cases 4 and 5 identified by targeted next-generation sequencing, only TEK gene mutation (missense mutations at T401M) was also detected by whole-genome sequencing. Whole-genome sequencing also failed to reveal mutations in any of the well-recognized intraductal papillary mucinous neoplasm or pancreatic ductal adenocarcinoma genes.
Discussion
Recent studies have helped to better characterize the histologic subtypes of intraductal papillary mucinous neoplasm15, 24, 32, 55, 56, 57, 58, 59, 60 and have confirmed that they have differences in genetic progression patterns compared with conventional pancreatic ductal adenocarcinoma.19, 30, 61, 62, 63 However, a focused study of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm has not been reported to date. Our results show that typical ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms do not harbor previously reported intraductal papillary mucinous neoplasm-related mutations (KRAS, GNAS, PIK3CA, CDKN2A/p16, SMAD4, TP53, etc.). Only one of our typical intraductal papillary mucinous neoplasms (Case 2) had an RNF43 mutation, which is a different mutation than previously reported intraductal papillary mucinous neoplasm-related RNF43 mutations.
Our results provide support for the proposition that ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm is a distinct entity with not only different morphologic features and biologic behavior, but also a different genotype. The most important genotypic difference from other subtypes of intraductal papillary mucinous neoplasm is the lack of KRAS mutations. KRAS gene mutations are frequent in intraductal papillary mucinous neoplasms without oncocytic differentiation. For example, Jang et al.22 investigated 37 intraductal papillary mucinous neoplasms and found mutations in codons 12 and 13 of the KRAS gene in 50% of pancreatobiliary subtype, 36% of gastric subtype, and of 21% of intestinal subtype. A review of relevant literature provided in their manuscript also shows that KRAS gene mutations were reported in up to 100% of intraductal papillary mucinous neoplasms.22 Similarly, in a prior study, our group identified mutations in codons 12 and 13 of the KRAS gene in 18 of 26 (69%) intraductal papillary mucinous neoplasm cases of other subtypes although there were no KRAS mutations in eight ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm cases.34 Patel et al.31 also found no activating point mutations in codons 12 and 13 of the KRAS gene in a single case of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm with associated invasive carcinoma. With possible evidence to the contrary, Xiao et al.35 identified somatic KRAS gene mutations in codon 12 in 3 of 18 (17%) ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms. However, the authors acknowledged they included ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms exhibiting heterogeneous epithelium.35 Therefore, it is quite possible that the three cases they reported as KRAS mutated may have exhibited heterogeneous epithelium, for which the oncocytic features were a morphologic variation within a non-oncocytic intraductal papillary mucinous neoplasm. As evidenced by our Case 11, the genotype of the intestinal and oncocytic components manifested similar mutations that are more typical of other intraductal papillary mucinous neoplasm subtypes.
We found further support of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm as an entity distinct from other subtypes of intraductal papillary mucinous neoplasm by the absence of GNAS mutations. Only Case 11 with heteregenous epithelium and an associated invasive colloid carcinoma manifested a GNAS mutation in all its components. GNAS gene mutations are common in other subtypes of intraductal papillary mucinous neoplasm, particularly in the intestinal subtype. Dal Molin et al.23 reported that 100% of intestinal subtype, 71% of pancreatobiliary subtype, and 51% of gastric subtype intraductal papillary mucinous neoplasms harbored a codon 201 GNAS mutation. The authors also analyzed two ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm cases, which were found to be GNAS wild type,23 in accordance with our findings. Similarly, in a recent study, our group has identified frequent GNAS gene mutations in non-oncocytic intraductal papillary mucinous neoplasms.26 Of note, no GNAS mutations have been identified in other pancreatic cystic neoplasms; nor were they identified in pancreatic ductal adenocarcinoma, suggesting that GNAS mutations are specific for the non-oncocytic intraductal papillary mucinous neoplasm phenotype.15, 23, 25
The lack of involvement in ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms of the typical genetic underpinnings that occur in other subtypes of intraductal papillary mucinous neoplasm should not come as a surprise considering the distinctive pathologic manifestations of this tumor type. Also, it is becoming clear in other organs that oncocytic neoplasms characterized by abundant mitochondrial accumulation have different identity than their non-oncocytic counterparts of the respective organs.12, 13 Similarly, the absence of the genetic alterations that underlie pancreatic ductal adenocarcinoma may explain the incomparably better clinical behavior of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms.1, 9 More importantly, these differences suggest that pancreatic ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm has distinct pathways of tumor progression, possibly through different underlying mechanisms causing different genetic alterations. In fact, the majority of the pancreatic cystic neoplasms (intraductal papillary mucinous neoplasms, mucinous cystic neoplasms, serous cyst adenomas and solid-pseudopapillary neoplasms) are associated with recurrent mutations in components of ubiquitin-dependent pathways.15 However, ‘oncocytic subtype’ of intraductal papillary mucinous neoplasm does not seem to be associated with ubiquitin-dependent pathways. In contrast, four specific genes (ARHGAP26, ASXL1, EPHA8, and ERBB4) were found to be mutated by targeted next-generateion sequencing in more than one of our cases (due to smaller sampling of target regions with whole-genome sequencing, compared to targeted next-generation sequencing, whole-genome sequencing may not detect variants of low allelic frequency). Excluding reports of potential involvement of the EPHA8 gene in pancreatic carcinogenesis,64, 65, 66, 67 these genes have not otherwise been previously associated with intraductal papillary mucinous neoplasms. Therefore, they are worth further scrutiny as being more than mere epiphenomena for their potential role in tumorigenesis of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms.
The ARHGAP26 gene encodes Rho GTPase activating protein 26 (ARHGAP26), also known as GTPase regulator associated with focal adhesion kinase (GRAF), in humans. It is recognized as a tumor suppressor gene that binds to focal adhesion kinase. Mutations and deletions of the GRAF gene are strongly implicated in leukemia development.68, 69, 70 The GRAF gene has also recently been shown to be mutated in gastric cancer.71, 72 The function of additional sex comb-like 1 (ASXL1) protein is not fully delineated,73 but it has been postulated that it may be involved in DNA and/or histone modification.74 Similar to ARHGAP26, mutations in ASXL1 have been identified in myelodysplastic syndromes75 and other myeloid malignancies, like acute myeloid leukemia, chronic myelomonocytic leukemia, and myeloproliferative neoplasia.76 Both EPHA8 and ERBB4 genes are members of human receptor tyrosine kinase family. EPHA8 encodes ephrin type-A receptor 8 protein. EPH and EPH-related receptors have been implicated in mediating developmental events, particularly in the nervous system.77 Genetic studies suggest that EPHA8 is involved in regulating cell adhesion78 and apoptosis.79 ERBB4 encodes receptor tyrosine-protein kinase erbB-4 enzyme, a member of the epidermal growth factor receptor subfamily. Mutations in this gene have been associated with diverse cancers including esophagus,80 gallbladder,81 and colon.82
In conclusion, although its intraductal nature and somewhat overlapping clinicopathologic features have led to classification of ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms as a variant of intraductal papillary mucinous neoplasm, our results suggest that, when defined strictly, ‘oncocytic subtype’ of intraductal papillary mucinous neoplasms are genetically distinct and thus should be recognized separately. Further analysis of molecular alterations in biologically distinct pathway(s) including ARHGAP26, ASXL1, EPHA8, and ERBB4 genes will likely shed new light on the mechanisms of intraductal tumor formation in the pancreas.
References
Adsay NV, Adair CF, Heffess CS et al. Intraductal oncocytic papillary neoplasms of the pancreas. Am J Surg Pathol 1996;20:980–994.
Adsay NV, Kloppel G, Fukushima N et al. Intraductal neoplasms of the pancreas. In: Bosman FT, Carneiro F, Hruban RH (eds). WHO Classification of Tumors. Lyon, France: WHO Press, 2010, pp 304–313.
Jyotheeswaran S, Zotalis G, Penmetsa P et al. A newly recognized entity: intraductal ‘oncocytic’ papillary neoplasm of the pancreas. Am J Gastroenterol 1998;93:2539–2543.
Adsay NV, Longnecker DS, Klimstra DS . Pancreatic tumors with cystic dilatation of the ducts: intraductal papillary mucinous neoplasms and intraductal oncocytic papillary neoplasms. Semin Diagn Pathol 2000;17:16–30.
D'Angelica M, Brennan MF, Suriawinata AA et al. Intraductal papillary mucinous neoplasms of the pancreas: an analysis of clinicopathologic features and outcome. Ann Surg 2004;239:400–408.
Sohn TA, Yeo CJ, Cameron JL et al. Intraductal papillary mucinous neoplasms of the pancreas: an updated experience. Ann Surg 2004;239:788–797; discussion 797–799.
Crippa S, Fernandez-Del Castillo C, Salvia R et al. Mucin-producing neoplasms of the pancreas: an analysis of distinguishing clinical and epidemiologic characteristics. Clin Gastroenterol Hepatol 2010;8:213–219.
Kloppel G, Basturk O, Schlitter AM et al. Intraductal neoplasms of the pancreas. Semin Diagn Pathol 2014;31:452–466.
Marchegiani G, Mino-Kenudson M, Ferrone CR et al. Oncocytic-type intraductal papillary mucinous neoplasms: a unique malignant pancreatic tumor with good long-term prognosis. J Am Coll Surg 2015;220:839–844.
Askan G, Klimstra DS, Adsay V et al. ‘Oncocytic-type’ of Intraductal Papillary Mucinous Neoplasm (IPMN): An Analysis of 25 Cases [abstract]. Laboratory Investigation 2016;96:1740.
Reid MD, Stallworth CR, Lewis MM et al. Cytopathologic diagnosis of oncocytic type intraductal papillary mucinous neoplasm: Criteria and clinical implications of accurate diagnosis. Cancer Cytopathol 2015;124:122–134.
Maximo V, Lima J, Soares P et al. Mitochondria and cancer. Virchows Arch 2009;454:481–495.
Maximo V, Lima J, Prazeres H et al. The biology and the genetics of Hurthle cell tumors of the thyroid. Endocr Relat Cancer 2012;19:R131–R147.
Fritz S, Fernandez-del Castillo C, Mino-Kenudson M et al. Global genomic analysis of intraductal papillary mucinous neoplasms of the pancreas reveals significant molecular differences compared to ductal adenocarcinoma. Ann Surg 2009;249:440–447.
Wu J, Jiao Y, Dal Molin M et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci USA 2011;108:21188–21193.
Furukawa T, Kuboki Y, Tanji E et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep 2011;1:161.
Schonleben F, Qiu W, Bruckman KC et al. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Lett 2007;249:242–248.
Z'Graggen K, Rivera JA, Compton CC et al. Prevalence of activating K-ras mutations in the evolutionary stages of neoplasia in intraductal papillary mucinous tumors of the pancreas. Ann Surg 1997;226:491–498; discussion 498–500.
Sessa F, Solcia E, Capella C et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Arch 1994;425:357–367.
Almoguera C, Shibata D, Forrester K et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988;53:549–554.
Kitago M, Ueda M, Aiura K et al. Comparison of K-ras point mutation distributions in intraductal papillary-mucinous tumors and ductal adenocarcinoma of the pancreas. Int J Cancer 2004;110:177–182.
Jang JY, Park YC, Song YS et al. Increased K-ras mutation and expression of S100A4 and MUC2 protein in the malignant intraductal papillary mucinous tumor of the pancreas. J Hepatobiliary Pancreat Surg 2009;16:668–674.
Dal Molin M, Matthaei H, Wu J et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol 2013;20:3802–3808.
Amato E, Molin MD, Mafficini A et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 2014;233:217–227.
Wu J, Matthaei H, Maitra A et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 2011;3:92ra66.
Tan MC, Basturk O, Brannon AR et al. GNAS and KRAS mutations define separate progression pathways in intraductal papillary mucinous neoplasm-associated carcinoma. J Am Coll Surg 2015;220:845–54.e1.
Jiang X, Hao HX, Growney JD et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2013;110:12649–12654.
Schonleben F, Qiu W, Ciau NT et al. PIK3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clin Cancer Res 2006;12:3851–3855.
Schonleben F, Qiu W, Remotti HE et al. PIK3CA, KRAS, and BRAF mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/C) of the pancreas. Langenbecks Arch Surg 2008;393:289–296.
Iacobuzio-Donahue CA, Klimstra DS, Adsay NV et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. Am J Pathol 2000;157:755–761.
Patel SA, Adams R, Goldstein M et al. Genetic analysis of invasive carcinoma arising in intraductal oncocytic papillary neoplasm of the pancreas. Am J Surg Pathol 2002;26:1071–1077.
Terris B, Dubois S, Buisine MP et al. Mucin gene expression in intraductal papillary-mucinous pancreatic tumours and related lesions. J Pathol 2002;197:632–637.
Mohri D, Asaoka Y, Ijichi H et al. Different subtypes of intraductal papillary mucinous neoplasm in the pancreas have distinct pathways to pancreatic cancer progression. J Gastroenterol 2012;47:203–213.
Chung SM, Hruban RH, Iacobuzio-Donahue C et al. Analysis of molecular alterations and differentiation pathways in intraductal oncocytic papillary neoplasm of the pancreas [abstract]. Mod Pathol 2005;18:277A–278A.
Xiao HD, Yamaguchi H, Dias-Santagata D et al. Molecular characteristics and biological behaviours of the oncocytic and pancreatobiliary subtypes of intraductal papillary mucinous neoplasms. J Pathol 2011;224:508–516.
Liszka L, Pajak J, Zielinska-Pajak E et al. Intraductal oncocytic papillary neoplasms of the pancreas and bile ducts: a description of five new cases and review based on a systematic survey of the literature. J Hepatobiliary Pancreat Sci 2010;17:246–261.
Won HH, Scott SN, Brannon AR et al. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp 2013;e50710.
Cheng DT, Cheng J, Mitchell TN et al. Detection of mutations in myeloid malignancies through paired-sample analysis of microdroplet-PCR deep sequencing data. J Mol Diagn 2014;16:504–518.
Cibulskis K, Lawrence MS, Carter SL et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219.
DePristo MA, Banks E, Poplin R et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
Robinson JT, Thorvaldsdottir H, Winckler W et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760.
McKenna A, Hanna M, Banks E et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303.
Wilm A, Aw PP, Bertrand D et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40:11189–11201.
Saunders CT, Wong WS, Swamy S et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–1817.
Ye K, Schulz MH, Long Q et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009;25:2865–2871.
Narzisi G, O'Rawe JA, Iossifov I et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods 2014;11:1033–1036.
Cingolani P, Platts A, Wang le L et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92.
Forbes SA, Bindal N, Bamford S et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39:D945–D950.
Xi R, Hadjipanayis AG, Luquette LJ et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci USA 2011;108:E1128–E1136.
Rausch T, Zichner T, Schlattl A et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28:i333–i339.
Wang J, Mullighan CG, Easton J et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 2011;8:652–654.
Chen K, Wallis JW, McLellan MD et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009;6:677–681.
Emde AK, Schulz MH, Weese D et al. Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics 2012;28:619–627.
Hruban RH, Takaori K, Klimstra DS et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol 2004;28:977–987.
Luttges J, Zamboni G, Longnecker D et al. The immunohistochemical mucin expression pattern distinguishes different types of intraductal papillary mucinous neoplasms of the pancreas and determines their relationship to mucinous noncystic carcinoma and ductal adenocarcinoma. Am J Surg Pathol 2001;25:942–948.
Nakamura A, Horinouchi M, Goto M et al. New classification of pancreatic intraductal papillary-mucinous tumour by mucin expression: its relationship with potential for malignancy. J Pathol 2002;197:201–210.
Yonezawa S, Horinouchi M, Osako M et al. Gene expression of gastric type mucin (MUC5AC) in pancreatic tumors: its relationship with the biological behavior of the tumor. Pathol Int 1999;49:45–54.
Adsay NV, Merati K, Basturk O et al. Pathologically and biologically distinct types of epithelium in intraductal papillary mucinous neoplasms: delineation of an ‘intestinal’ pathway of carcinogenesis in the pancreas. Am J Surg Pathol 2004;28:839–848.
Yonezawa S, Nakamura A, Horinouchi M et al. The expression of several types of mucin is related to the biological behavior of pancreatic neoplasms. J Hepatobiliary Pancreat Surg 2002;9:328–341.
Biankin AV, Biankin SA, Kench JG et al. Aberrant p16(INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut 2002;50:861–868.
Yamaguchi K, Chijiiwa K, Noshiro H et al. Ki-ras codon 12 point mutation and p53 mutation in pancreatic diseases. Hepatogastroenterology 1999;46:2575–2581.
Sato N, Fukushima N, Maitra A et al. Gene expression profiling identifies genes associated with invasive intraductal papillary mucinous neoplasms of the pancreas. Am J Pathol 2004;164:903–914.
Jones S, Zhang X, Parsons DW et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–1806.
Biankin AV, Waddell N, Kassahn KS et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405.
Murphy SJ, Hart SN, Lima JF et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013;145:1098–1109.e1.
Jiao Y, Yonescu R, Offerhaus GJ et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 2014;232:428–435.
Bojesen SE, Ammerpohl O, Weinhausl A et al. Characterisation of the GRAF gene promoter and its methylation in patients with acute myeloid leukaemia and myelodysplastic syndrome. Br J Cancer 2006;94:323–332.
Qian J, Qian Z, Lin J et al. Aberrant methylation of GTPase regulator associated with the focal adhesion kinase (GRAF) promoter is an adverse prognostic factor in myelodysplastic syndrome. Eur J Haematol 2010;85:174–176.
Qian Z, Qian J, Lin J et al. GTPase regulator associated with the focal adhesion kinase (GRAF) transcript was down-regulated in patients with myeloid malignancies. J Exp Clin Cancer Res 2010;29:111.
Kakiuchi M, Nishizawa T, Ueda H et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet 2014;46:583–587.
Wang K, Yuen ST, Xu J et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 2014;46:573–582.
Graubert T, Walter MJ . Genetics of myelodysplastic syndromes: new insights. Hematology Am Soc Hematol Educ Program 2011;2011:543–549.
Fisher CL, Randazzo F, Humphries RK et al. Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 2006;369:109–118.
Rocquain J, Carbuccia N, Trouplin V et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 2010;10:401.
Chen TC, Hou HA, Chou WC et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J 2014;4:e177.
Gu C, Shim S, Shin J et al. The EphA8 receptor induces sustained MAP kinase activation to promote neurite outgrowth in neuronal cells. Oncogene 2005;24:4243–4256.
Gu C, Park S . The EphA8 receptor regulates integrin activity through p110gamma phosphatidylinositol-3 kinase in a tyrosine kinase activity-independent manner. Mol Cell Biol 2001;21:4579–4597.
Kim Y, Park E, Noh H et al. Expression of EphA8-Fc in transgenic mouse embryos induces apoptosis of neural epithelial cells during brain development. Dev Neurobiol 2013;73:702–712.
Zhao K, Chen BJ, Chen ZG . ErbB4 as a potential molecular target in the treatment of esophageal squamous cell cancers. ScientificWorldJournal 2014;2014:124105.
Li M, Zhang Z, Li X et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat Genet 2014;46:872–876.
Williams CS, Bernard JK, Demory Beckler M et al. ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis 2015;36:710–718.
Acknowledgements
This work has been supported by a gift from the Farmer Family Foundation. We thank Ms. Dana Haviland and Ms. Tanisha Daniel for their assistance during manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This study was presented in part at the 103rd annual meeting of the United States and Canadian Academy of Pathology in Boston, MA, in March 2015.
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Basturk, O., Tan, M., Bhanot, U. et al. The oncocytic subtype is genetically distinct from other pancreatic intraductal papillary mucinous neoplasm subtypes. Mod Pathol 29, 1058–1069 (2016). https://doi.org/10.1038/modpathol.2016.98
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.98
This article is cited by
-
Preoperatively diagnosed intraductal oncocytic papillary neoplasm of the pancreas with prominent invasion: a case report
Clinical Journal of Gastroenterology (2023)
-
Intraductal oncocytic papillary neoplasm of the pancreas: clinical and radiological features compared to those of intraductal papillary mucinous neoplasm
Abdominal Radiology (2023)
-
Intraductal oncocytic papillary neoplasm arising in Peutz-Jeghers Syndrome bile duct: a unique case report
Diagnostic Pathology (2022)
-
Pancreatic ductal adenocarcinomas associated with intraductal papillary mucinous neoplasms (IPMNs) versus pseudo-IPMNs: relative frequency, clinicopathologic characteristics and differential diagnosis
Modern Pathology (2022)
-
Investigation of -PRKACA/-PRKACB fusion genes in oncocytic tumors of the pancreatobiliary and other systems
Virchows Archiv (2022)
