
Abstract
Kinase activation by chromosomal translocations is a common mechanism that drives tumorigenesis in spitzoid neoplasms. To explore the landscape of fusion transcripts in these tumors, we performed whole-transcriptome sequencing using formalin-fixed, paraffin-embedded (FFPE) tissues in malignant or biologically indeterminate spitzoid tumors from 7 patients (age 2–14 years). RNA sequence libraries enriched for coding regions were prepared and the sequencing was analyzed by a novel assembly-based algorithm designed for detecting complex fusions. In addition, tumor samples were screened for hotspot TERT promoter mutations, and telomerase expression was assessed by TERT mRNA in situ hybridization (ISH). Two patients had widespread metastasis and subsequently died of disease, and 5 patients had a benign clinical course on limited follow-up (mean: 30 months). RNA sequencing and TERT mRNA ISH were successful in six tumors and unsuccessful in one disseminating tumor because of low RNA quality. RNA sequencing identified a kinase fusion in five of the six sequenced tumors: TPM3–NTRK1 (2 tumors), complex rearrangements involving TPM3, ALK, and IL6R (1 tumor), BAIAP2L1–BRAF (1 tumor), and EML4–BRAF (1 disseminating tumor). All predicted chimeric transcripts were expressed at high levels and contained the intact kinase domain. In addition, two tumors each contained a second fusion gene, ARID1B–SNX9 or PTPRZ1–NFAM1. The detected chimeric genes were validated by home-brew break-apart or fusion fluorescence in situ hybridization (FISH). The two disseminating tumors each harbored the TERT promoter –124C>T (Chr 5:1,295,228 hg19 coordinate) mutation, whereas the remaining five tumors retained the wild-type gene. The presence of the –124C>T mutation correlated with telomerase expression by TERT mRNA ISH. In summary, we demonstrated complex fusion transcripts and novel partner genes for BRAF by RNA sequencing of FFPE samples. The diversity of gene fusions demonstrated by RNA sequencing defines the molecular heterogeneity of spitzoid neoplasms.
Similar content being viewed by others

Main
Spitzoid tumors are a clinicopathologically distinct class of melanocytic neoplasms that occur more commonly in younger individuals and account for the majority of so-called ‘melanomas’ seen in the pediatric population. Histologically, these lesions are characterized by compound or dermal proliferations of large epithelioid and/or spindle-shaped melanocytes having abundant eosinophilic cytoplasm, often forming junctional nests in conjunction with epidermal hyperplasia. The lack of objective criteria to determine the malignant potential of spitzoid tumors is a major diagnostic challenge.1, 2, 3, 4, 5 The established histopathologic criteria used to differentiate nevi from conventional melanoma are not reliable for spitzoid neoplasms. Also, unlike conventional melanoma, lymph node metastasis in general is not predictive of poor clinical outcome in patients with spitzoid tumors.6, 7, 8, 9, 10, 11, 12, 13
Spitzoid lesions with features significantly deviating from a stereotypical Spitz nevus, such as large lesional size (>1 cm), asymmetry, ulceration, pagetoid melanocytosis extending peripherally, significant intradermal mitotic activity, lack of cellular maturation with depth, confluent cellular growth, involvement of subcutaneous fat, or severe cytologic atypia, are considered atypical spitzoid melanocytic proliferations, encompassing atypical Spitz tumor and spitzoid melanoma. Additional important clinical information includes the age of the patient, as melanoma is extremely rare under the age of 10 years, and clinical features such as a new or rapidly growing lesion, asymmetry, irregular coloration, ulceration, bleeding, and history of trauma. The diagnosis of spitzoid melanoma in these circumstances is considered when multiple chromosomal aberrations are detected by using ancillary molecular techniques, such as the multiprobe fluorescent in situ hybridization (FISH) assay14, 15 or comparative genomic hybridization analysis.16, 17 The true predictive value of these assays for determining clinical outcome in spitzoid tumors, however, remains uncertain. We recently evaluated 56 spitzoid tumors for the presence of telomerase reverse transcriptase (TERT) promoter mutations and their association with disease progression. We found a hotspot TERT promoter mutation in tumors from patients who had a malignant clinical course but not in tumors from patients who had a favorable clinical outcome, suggesting that these mutations contribute to malignant biological behavior.12 Nonetheless, the underlying molecular mechanisms responsible for the potential of these lesions to spread distantly need to be investigated further.
The Cancer Genome Atlas Network has recently proposed a genomic classification of cutaneous melanomas into four mutually exclusive genetic subtypes on the basis of the presence of a hotspot mutation in the significantly mutated melanoma-associated genes, BRAF, RAS (N/K/H), NF1, and the triple wild-type.18 By this stratification scheme, most spitzoid melanomas are likely to fall into the triple wild-type subtype,19, 20 a heterogeneous molecular category shown to be enriched by focal amplifications or complex structural rearrangements.18 Wiesner et al. and others21, 22, 23, 24 demonstrated that instead of activation of the MAP kinase pathway through point mutations, chromosomal translocation-induced kinase fusions drive tumorigenesis in spitzoid neoplasms. These rearrangements are predicted to constitutively activate the MAP kinase pathway by an in-frame fusion of the receptor tyrosine kinase NTRK1, ROS1, RET, ALK, or MET or the serine/threonine kinase BRAF to the N terminal of various 5′ partner genes.21, 23, 24 As these genetic alterations are present in the entire biologic spectrum of the disease, that is, the benign (nevi), the biologically indeterminate or low-grade malignant (atypical Spitz tumors), and the overtly malignant lesions (spitzoid melanoma), they are likely acquired in the early stage of disease but cannot by themselves lead to melanoma.25, 26
To explore the landscape of structural rearrangements in spitzoid melanomas, in the current study we used RNA sequencing to characterize the transcriptome of seven histologically malignant or biologically indeterminate spitzoid tumors. Furthermore, we used TERT mRNA in situ hybridization (ISH) to demonstrate the association between TERT promoter mutations and telomerase expression at the cellular level.
Materials and methods
Study Population
The study was approved by the institutional review boards of participating institutions. The study subjects were selected from a previously reported cohort of 56 patients with spitzoid melanocytic tumors12 for whom documented clinical outcomes and sufficient biological material were available. To improve the performance of RNA sequencing, only biological samples with a storage time of ≤7 years were considered for the study. As an exception, an old archived formalin-fixed, paraffin-embedded (FFPE) block (>20 years old) from a rare fatal spitzoid melanoma in a young patient was also included. Adequate biologic material was obtained for RNA sequencing from seven malignant or biologically indeterminate spitzoid tumors (five primary tumors and two metastatic tumors).
The hotspot BRAF, NRAS, and TERT promoter mutation data on these tumors have been previously reported.12 In summary, genomic DNA was extracted from the tumor samples (five primary tumors, one paired primary and metastatic tumor, and one metastatic tumor) and screened for hotspot mutations of the genes by PCR and Sanger sequencing, as previously described.20
Transcriptome Sequencing
Tumor tissue samples from 8 to 10 FFPE slide-mounted sections were manually dissected, with corresponding H&E sections used to guide dissections, to obtain at least 70% tumor purity. RNA was isolated by using the Maxwell system (Promega). RNA was quantitated by fluorescence dye staining by using the Quant-iT (Life Technologies) RNA assay. RNA quality was evaluated by using a 2100 Bioanalyzer (Agilent Technologies) with a Nano RNA 6000 Chip. RNASEQ libraries enriched for coding regions were prepared by using the Truseq RNA Access Library Prep Kit (Illumina), following the manufacturer’s protocol for RNA input quantity relative to RNA quality. Sequencing was performed on HiSeq2000 (Illumina) to generate 100-bp paired-end reads.
RNA Sequencing Analysis
RNA sequencing data were generated as previously described.25 Paired-end reads from RNA sequences were aligned to the following 4 database files by using BWA (0.5.10) aligner: (1) the human GRCh37-lite reference sequence, (2) RefSeq, (3) a sequence file representing all possible combinations of nonsequential pairs in RefSeq exons, and (4) an AceView database flat file downloaded from UCSC, representing transcripts constructed from human expressed sequence tags. The mapping results from files 2, 3, and 4 were aligned to human reference genome coordinates and also to the human GRCh37-lite reference sequence by using STAR 2.3.0 without annotations. The final BAM file was constructed by selecting the best alignment among the five mappings. The coverage was calculated by using an in-house pipeline. Structural variations were detected by using CICERO, a novel algorithm that uses de novo assembly to identify structural variations in RNA sequences.
Fluorescence In Situ Hybridization
BAC clones (BACPAC Resources) were used to develop break-apart probes for the following genes: BRAF (RP11-837G3, RP11-948O19), NTRK1 (CH17-67O18, RP11-1038N13), PTPRZ1 (CH17-132B19, RP11-99L10), IL6R (CH17-169C19, RP11-627K14), TPM3 (CH17-317C21, CH17-169C19), EML4 (CH17-315G08, RP11-885P15), ARID1B (RP11-230C9, CH17-280H05), and BAIAP2L1 (RP11-958G24, CH17-112O19). Break-apart FISH for ALK was performed by using a commercially available probe set (CytoCell, Cat. no. LPS 019-A). In addition, BAC clones (CH17-132B19, RP11-99L10 and CH17-57M15, CH17-240N01) were used to develop a fusion probe set for PTPRZ1–NFAM1. Dual-color FISH was performed on 4 μm FFPE sections, as previously described.20
TERT mRNA ISH
The mRNA ISH, a novel method to detect mRNA in FFPE tissues,26 was performed for TERT mRNA on a Discovery Ultra automation system (Ventana Medical Systems) by using RNAscope VS Reagent Kit–RED (Advanced Cell Diagnostics). VS Probe–Hs-TERT (Cat. no.605516) specific to the sequence region between nucleotides 2164 and 3231 encoding the TERT transcript was used according to the manufacturer’s instructions. Briefly, 4 μm FFPE tissue sections of tumors were pretreated in citrate buffer with heat, followed by protease digestion before hybridization with the target oligo probes. Slides were hybridized sequentially with target probes incubated at 43 °C for 2 h and 32 min, preamplifier at 53 °C for 32 min, and amplifier at 53 °C for 32 min, and label probes at room temperature for 12 min. Between the hybridization steps, slides were washed with Ribowash buffer (0.1 × saline sodium citrate). Hybridization signals were detected by chromogenic development with Fast Red, followed by counterstaining with hematoxylin. Each sample was quality controlled for RNA integrity with an RNAscope probe for PPIB RNA and for background with a probe for bacterial dapB RNA. The specific RNA staining signal was identified as intracellular red punctate dots.
Results
Clinicopathologic Findings
Table 1 and Figures 1, 2, 3, 4 and 5 show the clinical and disease characteristics for the seven patients. Tumors were detected in samples from 4 children (age 2–7 years) and 3 adolescents (age 11–14 years) and involved the lower extremities (n=5), ear (n=1), and trunk (n=1). Two patients had clinically detectable lymphadenopathy (macrometastasis). Of the 6 patients whose sentinel/regional lymph nodes were examined, 5 patients were positive for nodal metastasis (Table 1), with clinically detectable lymphadenopathy (patients 5 and 7), large nodal deposits (patients 2 and 3), and isolated tumor cells (patient 1). At a median follow-up of 20 months (range, 6–72 months), 5 patients were alive and well with no evidence of disease and 2 patients developed distant metastasis in the lungs and brain and subsequently succumbed to the disease 18 and 24 months after diagnosis (Table 1).

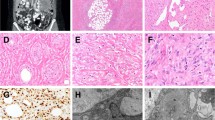
Spitzoid melanoma with the EML4–BRAF fusion transcript. Analysis of a tumor sample from a 14 year-old male (patient 1) with a 1.3-mm-thick spitzoid melanoma with microscopic sentinel lymph node metastasis at diagnosis and death as a result of disseminated disease in 18 months. (a, b) H&E photomicrographs (10 × and 40 ×) of the primary tumor show compound proliferation of epithelioid and spindle-shaped spitzoid melanocytes arranged in confluent junctional nests with ulceration. (c) TERT mRNA ISH shows bright intracellular signals in melanocytes in this TERT promoter mutant melanoma. (d, e) Break-apart FISH shows splits of the red and green signals, consistent with the rearrangement of BRAF and EML4, respectively. (f) Schematic figure depicting the reciprocal translocation between 2p21 and 7q34, resulting in the chimeric products EML4–BRAF and BRAF–EML4. (g) The in-frame fusion transcript produced by joining exon 6 of EML4 (NM_019063, chr2:42491872) to exon 10 of BRAF (NM_004333, chr7:140482957). (h) The fusion transcript results in a 596-amino-acid chimeric protein containing the intact kinase domain of BRAF. (i) The RNA transcript contig shows the fusion breakpoint in the EML4–BRAF chimeric gene.


Spitzoid neoplasm with the BAIAP2L1–BRAF fusion transcript. Analysis of a tumor sample from a 4-year-old African-American female (patient 2) with a 7.8-mm-thick spitzoid melanocytic proliferation on her thigh, sentinel lymph node metastasis at diagnosis, and no evidence of disease at 53 months of follow-up. (a) H&E photomicrograph (4 ×) showing a dome-shaped proliferation of vertically oriented fascicles of spindled melanocytes throughout the reticular dermis. (b) TERT mRNA ISH shows no detectable intracellular signals beyond the background expression in this wild-type TERT promoter melanocytic tumor. (c) Break-apart FISH shows the rearrangement of BRAF with loss of the 5′ end (red signal), consistent with a complex translocation. (d) Break-apart FISH for BAIAP2L1 shows splits of the red and green signals. (e) Schematic representation of the BAIAP2L1–BRAF fusion, showing that the fusion occurred by tandem duplication at 7q21.3-7q34. (f) The fusion transcript is formed by joining exon 12 of BAIAP2L1 (NM_018842, chr7:97933507) to exon 9 of BRAF (NM_004333, chr7:140487384). (g) The fusion product is an 860-amino-acid chimeric protein containing the entire kinase domain of BRAF. (h) The RNA transcript contig shows the fusion breakpoint in the chimeric BAIAP2L1-BRAF transcript.

Multiple isoforms of the TPM3–NTRK1 fusion transcripts in two spitzoid melanocytic tumors. (a) Photograph of a 2-year-old boy (patient 3) with an amelanotic exophytic nodule on the thigh that was clinically thought to be a pyogenic granuloma. (b) H&E photomicrograph (4 ×) of the upper part of the lesion shows fascicles of spindled and epithelioid melanocytes arranged in whorls and nests with edema of the papillary dermis with telangiectasia. There were lymphovascular invasion and focal necrosis (not shown). (c) H&E photomicrograph (4 ×) of the bottom part of the lesion shows dumbbell-shaped configuration and deep extension into the subcutaneous fat. (d, f) H&E photomicrographs (4 ×) of the top and bottom portions of a lesion on the ear of a 6-year-old female (patient 5) showing nests of polygonal-shaped epithelioid melanocytes at high magnification (inset in e) that extend throughout the dermis and far into the basal line of resection margin in subcutis. (g, h) Break-apart FISH for NTRK1 shows split signals in both spitzoid tumors. (i–k) The NTRK1 fusion gene encoded two variant TPM3–NTRK1 isoforms in patient 3 and a third isoform in patient 5. All isoforms retained the intact tyrosine kinase domain of NTRK1.

Complex genomic rearrangements involving ALK, TPM3, and IL6R. Spitzoid melanocytic neoplasm in the calf of a 13-year-old female (patient 4). (a) H&E photomicrograph showing nests of spindled melanocytes with ulceration. (b) ALK immunoreactivity highlights a fascicular architecture in the dermis. (c, d) Break-apart FISH for ALK and IL6R confirms rearrangements in the respective genes. (e–h) Schematic of the TPM3–ALK fusion gene. The first 222 amino acids of TPM3 are fused to the C terminus of ALK, incorporating the intact tyrosine kinase domain of ALK. The remaining TPM3 protein is fused to the IL6R protein. The identification of multiple fusions involving TPM3 suggests a complex translocation event.

Spitzoid melanoma with a TERT promoter mutation and fatal outcome. An 11-year-old girl (patient 7) presented with a lesion on her thigh with an initial histologic diagnosis of a Spitz nevus, bulky nodal metastasis in 6 months, and disseminated disease and death 24 months after diagnosis. (a) Scanning magnification shows closely packed nests of spindle-shaped melanocytes with pseudoepitheliomatous hyperplasia and telangiectasia in the upper dermis. (b) The lower half of the lesion shows dense cellularity, sheet-like arrangement, and deep extension into subcutis. (c) Intermediate magnification shows confluent fascicles of spindle-shaped melanocytes. (d) Metastatic lung nodule composed of morphologically similar spindled melanocytes.
BRAF, NRAS, and TERT Promoter Mutations
The seven spitzoid tumors were negative for the activating point mutations in BRAF and NRAS. The two tumors that led to a fatal outcome each harbored a TERT promoter mutation at –124 bp from the ATG start site (–124C>T) in the primary and metastatic samples, whereas the five other tumors retained the TERT promoter wild type (Table 1).
Fusion Transcripts by RNA Sequencing
RNA sequencing was successful in 6 of the 7 samples, with a minimum 20 × coverage of at least 20% exonic bases and a median coverage of 10 × in all RefSeq annotated exons (Supplementary Table 1). Coverage was low in one sample obtained from old FFPE material (patient 7), and it was excluded from analysis. RNA sequencing identified a kinase fusion in five of the six successfully tested tumors (Supplementary Table 2). The following fusion genes were identified by RNA sequencing: EML4–BRAF (1 disseminating tumor; Figure 1), BAIAP2L1–BRAF (1 tumor; Figure 2), TPM3–NTRK1 (2 tumors; Figure 3), and TPM3–ALK (1 tumor; Figure 4). All predicted chimeric transcripts were expressed at high levels and contained the intact kinase domain. The FPKM (fragment per kb per million mapped reads) expression values for the fusion genes are provided in Supplementary Table 2. In addition, two spitzoid tumors each carried a second fusion gene, ARID1B–SNX9 and PTPRZ1–NFAM1 (Supplementary Figure 1). There was no structural rearrangement in one of the successfully sequenced samples (patient 6).
Fluorescence In Situ Hybridization
Break-apart FISH for BRAF (2 tumors), NTRK1 (2 tumors), PTPRZ1 (1 tumor), IL6R (1 tumor), TPM3 (3 tumors), EML4 (1 tumor), ARID1B (1 tumor), and BAIAP2L1 (1 tumor) showed split signals in at least 30% of the evaluated cells, indicating rearrangement of the respective genes (Figures 1, 2, 3 and 4 and Supplementary Figure 1). The PTPRZ1–NFAM1 fusion FISH showed multiple copies of overlapping signals (Supplementary Figure 1), suggesting gene fusion followed by copy gain in the kinase fusion gene.
TERT mRNA ISH
TERT mRNA ISH showed distinct bright intracellular signals in melanocytes in the TERT promoter mutant metastasizing tumor (Figure 1) but not in the wild-type TERT promoter tumors (Figure 2). TERT mRNA ISH was not successful in one sample (patient 7) because of low RNA quality.
Discussion
By using RNA sequencing, we identified in-frame fusions of kinases, BRAF, NTRK1, and ALK, in a mutually exclusive pattern, with various partner genes in five of the six successfully sequenced spitzoid tumors. We found two novel 5′ BRAF fusion partners EML4 and BAIAP2L1, expanding the list of BRAF N-terminal fusion partners previously described in pilocytic astrocytoma and melanocytic tumors.21, 22, 23, 27 EML4 is a recurrent fusion partner gene with ALK, and the resulting fusion transcript EML4–ALK is the primary oncogenic driver in 3–6% of non-small-cell lung carcinomas.28, 29 However, to our knowledge, there are no reports of EML4 participating in an oncogenic fusion with BRAF. BAIAP2L1 (BAI1-associated protein 2-like 1) has been reported to participate in the fusion transcript with FGFR3 in bladder and lung cancer, but has never been described previously in melanoma.30, 31, 32
One of the samples in our study (patient 4) harbored the TPM3–ALK fusion that has been previously reported in spitzoid neoplasms.33, 34 Interestingly, before the discovery of its oncogenic association with melanocytic neoplasms, TPM3–ALK was identified in tumors from other lines of differentiation, namely the mesenchymal (inflammatory myofibroblastic tumor),35 the lymphoid (anaplastic large-cell lymphoma),36 and the epithelial (squamous cell carcinoma and renal cancer) lineages.37, 38 This finding supports the assertion that known fusion genes can drive oncogenesis in tumors of different cell types. In addition to translocations, ALK is also activated through a de novo alternative transcription initiation, without genetic alterations at the ALK locus, in ∼11% of melanomas.39
Certain morphologic features have been linked to spitzoid tumors with ALK rearrangement.24, 34 Similar features were seen in our patient sample with ALK fusion that exhibited nests of spindle-shaped melanocytes with fascicular infiltration of the dermis and subcutis (Figure 4). Although no definitive conclusions can be drawn because of the small number of samples in this study, the two samples with NTRK1 fusion shared a few morphologic features such as prominent cellularity, deep extension, nested arrangement in the upper part, and confluent cellular nodules with rounded pushing margins at the bottom (Figure 3). How reliably these morphologic manifestations predict the type of fusion transcripts has not yet been sufficiently studied. In any case, given the diversity of gene fusions involving alternative partners or even alternative exons within the same pairs of genes, as demonstrated in two spitzoid melanomas in this series harboring TMP3–NTRK1 (Figure 3), or other coexisting genetic alterations, the morphologic heterogeneity in spitzoid tumors even with the same kinase fusion is not unexpected.
Of the two patients with fatal outcomes in our study, one carried a BRAF fusion and the sample from the other patient could not be successfully sequenced because of degraded RNA. The second patient with BRAF fusion in our series had a benign course of disease. To date, the association between the type of fusion gene and prognosis in patients with spitzoid tumors remains uncertain. Tumors from both patients with fatal outcomes harbored the –124C>T transcriptional activating mutation in the TERT promoter. The –124C>T mutation (also referred to as C228T in the literature) has been previously shown to correlate with high TERT mRNA expression in melanoma,18 and here on a different platform using TERT mRNA ISH, we demonstrated its association with telomerase expression at a cellular level (Figure 1).
Transcriptome sequencing identified additional novel fusion genes accompanied with kinase fusions in spitzoid tumors. One sample harbored the PTPRZ1–NFAM1 that was associated with elevated expression of the NFAT activated protein with ITAM motif 1 (NFAM1) (FPKM=621.2). The protein encoded by NFAM1 contains an immunoreceptor tyrosine-based activation motif that is thought to regulate the development of B cells,40 but its role in cancer development and melanoma is not well studied. The 5′ partner gene PTPRZ1 is a recurrent fusion partner with MET in glioblastoma.41 The fusion transcript ARID1B–SNX9 is expected to lead to the loss of function of the tumor suppressor gene ARID1B, a subunit of the SWI/SNF complex. Although the exact function of ARID1B in melanoma has not been investigated, 13% of melanomas have a loss-of-function mutation in a component of the SWI/SNF complex,42 suggesting that the chromatin remodeling complex plays a role in melanoma tumorigenesis. Although the oncogenic contribution of these genetic alterations remains speculative until they are functionally characterized, our findings, together with the complex nature of translocation events seen in the tumor samples, suggest that spitzoid tumors are enriched with structural rearrangements.
Notably, no structural rearrangement was identified in one spitzoid tumor in our study (Table 1; patient 6), even though the exonic coverage for this sample was comparable to that of other specimens (Supplementary Table 1). Therefore, we speculate that in a subset of spitzoid neoplasms, mechanisms other than translocations can activate oncogenes. Whole-genome sequencing can give further insights into the oncogenic mechanisms of these tumors.
In summary, we demonstrate complex and heterogeneous structural rearrangements in spitzoid tumors by transcriptome sequencing by using FFPE tissue. The heterogeneity of the fusion transcripts observed by RNA sequencing correlates with the morphologic and clinical diversity of this group of melanocytic tumors. The association between TERT promoter mutations or telomerase expression with outcomes in patients with spitzoid melanocytic tumors requires further studies.
References
Reed D, Kudchadkar R, Zager JS et al. Controversies in the evaluation and management of atypical melanocytic proliferations in children, adolescents, and young adults. J Natl Compr Canc Netw 2013;11:679–686.
Barnhill RL, Argenyi ZB, From L et al. Atypical Spitz nevi/tumors: lack of consensus for diagnosis, discrimination from melanoma, and prediction of outcome. Hum Pathol 1999;30:513–520.
Gerami P, Busam K, Cochran A et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol 2014;38:934–940.
Spatz A, Barnhill RL . The Spitz tumor 50 years later: revisiting a landmark contribution and unresolved controversy. J Am Acad Dermatol 1999;40:223–228.
Cerroni L, Barnhill R, Elder D et al. Melanocytic tumors of uncertain malignant potential: results of a tutorial held at the XXIX Symposium of the International Society of Dermatopathology in Graz, October 2008. Am J Surg Pathol 2010;34:314–326.
Paradela S, Fonseca E, Pita S et al. Spitzoid melanoma in children: clinicopathological study and application of immunohistochemistry as an adjunct diagnostic tool. J Cutan Pathol 2009;36:740–752.
Berk DR, LaBuz E, Dadras SS et al. Melanoma and melanocytic tumors of uncertain malignant potential in children, adolescents and young adults—the Stanford experience 1995-2008. Pediatr Dermatol 2010;27:244–254.
Busam KJ, Pulitzer M . Sentinel lymph node biopsy for patients with diagnostically controversial Spitzoid melanocytic tumors? Adv Anat Pathol 2008;15:253–262.
Hung T, Piris A, Lobo A et al. Sentinel lymph node metastasis is not predictive of poor outcome in patients with problematic spitzoid melanocytic tumors. Hum Pathol 2013;44:87–94.
Ludgate MW, Fullen DR, Lee J et al. The atypical Spitz tumor of uncertain biologic potential: a series of 67 patients from a single institution. Cancer 2009;115:631–641.
Lallas A, Kyrgidis A, Ferrara G et al. Atypical Spitz tumours and sentinel lymph node biopsy: a systematic review. Lancet Oncol 2014;15:e178–e183.
Lee S, Barnhill RL, Dummer R et al. TERT promoter mutations are predictive of aggressive clinical behavior in patients with spitzoid melanocytic neoplasms. Sci Rep 2015;5:11200.
Busam KJ, Murali R, Pulitzer M et al. Atypical spitzoid melanocytic tumors with positive sentinel lymph nodes in children and teenagers, and comparison with histologically unambiguous and lethal melanomas. Am J Surg Pathol 2009;33:1386–1395.
Gerami P, Zembowicz A . Update on fluorescence in situ hybridization in melanoma: state of the art. Arch Pathol Lab Med 2011;135:830–837.
Gerami P, Scolyer RA, Xu X et al. Risk assessment for atypical spitzoid melanocytic neoplasms using FISH to identify chromosomal copy number aberrations. Am J Surg Pathol 2013;37:676–684.
Bastian BC, Olshen AB, LeBoit PE et al. Classifying melanocytic tumors based on DNA copy number changes. Am J Pathol 2003;163:1765–1770.
Ali L, Helm T, Cheney R et al. Correlating array comparative genomic hybridization findings with histology and outcome in spitzoid melanocytic neoplasms. Int J Clin Exp Pathol 2010;3:593–599.
Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015;161:1681–1696.
Yazdan P, Cooper C, Sholl LM et al. Comparative analysis of atypical spitz tumors with heterozygous versus homozygous 9p21 deletions for clinical outcomes, histomorphology, BRAF mutation, and p16 expression. Am J Surg Pathol 2014;38:638–645.
Lu C, Zhang J, Nagahawatte P et al. The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol 2015;135:816–823.
Wiesner T, He J, Yelensky R et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014;5:3116.
Hutchinson KE, Lipson D, Stephens PJ et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 2013;19:6696–6702.
Botton T, Yeh I, Nelson T et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res 2013;26:845–851.
Yeh I, Botton T, Talevich E et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 2015;6:7174.
Parker M, Mohankumar KM, Punchihewa C et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014;506:451–455.
Wang F, Flanagan J, Su N et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 2012;14:22–29.
Jones DT, Kocialkowski S, Liu L et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008;68:8673–8677.
Kwak EL, Bang YJ, Camidge DR et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693–1703.
Takeuchi K, Choi YL, Soda M et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res 2008;14:6618–6624.
Williams SV, Hurst CD, Knowles MA . Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet 2013;22:795–803.
Nakanishi Y, Akiyama N, Tsukaguchi T et al. Mechanism of oncogenic signal activation by the novel fusion kinase FGFR3-BAIAP2L1. Mol Cancer Ther 2015;14:704–712.
Shinmura K, Kato H, Matsuura S et al. A novel somatic FGFR3 mutation in primary lung cancer. Oncol Rep 2014;31:1219–1224.
Yeh I, de la Fouchardiere A, Pissaloux D et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581–591.
Busam KJ, Kutzner H, Cerroni L et al. Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am J Surg Pathol 2014;38:925–933.
Lawrence B, Perez-Atayde A, Hibbard MK et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377–384.
Lamant L, Dastugue N, Pulford K et al. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999;93:3088–3095.
Jazii FR, Najafi Z, Malekzadeh R et al. Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J Gastroenterol 2006;12:7104–7112.
Sugawara E, Togashi Y, Kuroda N et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer 2012;118:4427–4436.
Wiesner T, Lee W, Obenauf AC et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015;526:453–457.
Ohtsuka M, Arase H, Takeuchi A et al. NFAM1, an immunoreceptor tyrosine-based activation motif-bearing molecule that regulates B cell development and signaling. Proc Natl Acad Sci USA 2004;101:8126–8131.
Bao ZS, Chen HM, Yang MY et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res 2014;24:1765–1773.
Hodis E, Watson IR, Kryukov GV et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–263.
Acknowledgements
This study was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number P30CA021765 and by ALSAC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work was presented in part at the 2015 United States and Canadian Academy of Pathology Annual Meeting in Boston, MA.
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Wu, G., Barnhill, R., Lee, S. et al. The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod Pathol 29, 359–369 (2016). https://doi.org/10.1038/modpathol.2016.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.37
This article is cited by
-
MITF::CREM-rearranged tumor: a novel group of cutaneous tumors with melanocytic differentiation
Virchows Archiv (2023)
-
Multiomic analysis of papillary thyroid cancers identifies BAIAP2L1-BRAF fusion and requirement of TRIM25, PDE5A and PKCδ for tumorigenesis
Molecular Cancer (2022)
-
Clinical, morphologic, and genomic findings in ROS1 fusion Spitz neoplasms
Modern Pathology (2021)
-
Spitz melanoma is a distinct subset of spitzoid melanoma
Modern Pathology (2020)
-
Frequent overexpression of klotho in fusion-negative phosphaturic mesenchymal tumors with tumorigenic implications
Modern Pathology (2020)
