
Abstract
We performed exome sequencing of 77 melanocytic specimens composed of Spitz nevi (n=29), Spitzoid melanomas (n=27), and benign melanocytic nevi (n=21), and compared the results with published melanoma sequencing data. Our study highlights the prominent similarity between Spitzoid and conventional melanomas with similar copy number changes and high and equal numbers of ultraviolet-induced coding mutations affecting similar driver genes. Mutations in MEN1, PRKAR1A, and DNMT3A in Spitzoid melanomas may indicate involvement of the protein kinase A pathway, or a role of DNA methylation in the disease. Other than activating HRAS variants, there were few additional mutations in Spitz nevi, and few copy number changes other than 11p amplification and chromosome 9 deletions. Similarly, there were no large-scale copy number alterations and few somatic alterations other than activating BRAF or NRAS mutations in conventional nevi. A presumed melanoma driver mutation (IDH1Arg132Cys) was revealed in one of the benign nevi. In conclusion, our exome data show significantly lower somatic mutation burden in both Spitz and conventional nevi compared with their malignant counterparts, and high genetic similarity between Spitzoid and conventional melanoma.
Similar content being viewed by others


Main
Recent exome sequencing studies have successfully uncovered several novel melanoma driver genes, including PPP6C, RAC1, and ARID2,1, 2, 3, 4, 5, 6 allowing for a better understanding of the pathways dysregulated in the disease. The results of these studies were also instrumental in classifying melanoma into four major subgroups, based on the mutational status of BRAF, RAS, and NF1.3, 7, 8
Although much has been learned about the genetic causes of melanoma, the molecular changes underlying benign melanocytic lesions are known to a lesser degree. For example, small sequencing studies were performed on specific nevi subtypes, including congenital nevi.9 In addition, a recent study interrogated the mutational status of 182 cancer-related genes in 30 Spitz nevi,10 a type of benign nevus with characteristic 11p amplifications, concurrent HRAS mutations, and distinct histopathological features. However, we are not aware of a large-scale, comprehensive, and unbiased genomic sequencing effort interrogating these lesions.
To address this, we exome-sequenced 29 Spitz nevi, 27 Spitzoid melanomas, and 21 conventional melanocytic nevi to compare two types of melanocytic neoplasm: (1) Spitz nevi to Spitzoid melanomas, because they share common histopathological features such as epithelioid or spindled melanocytes, and (2) conventional melanocytic nevi to conventional melanoma (non-Spitzoid, referred to as melanoma below), using data from a recent exome screen in melanoma.7 We hypothesized that a comprehensive exome-wide sequencing effort of these lesions should facilitate (a) a better understanding of the nevi mutational landscape, (b) insights into early mutational changes, and (c) a refined assessment of published melanoma driver genes by studying their presence or absence in nevi. Spitzoid lesions were of particular interest because there is an urgent need to discover markers that can accurately diagnose benign Spitz nevi present predominantly in children and young adults, and differentiate them from Spitzoid melanomas. The results of the studies can also address the question whether Spitz nevi and Spitzoid melanomas represent early and late stages of a distinct melanocytic subgroup with low UV exposure, as reported before.11
Materials and methods
Collection of Samples and Clinical Data
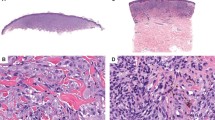
The 29 Spitz nevi and 27 Spitzoid melanomas were retrieved from the Yale Spitzoid Neoplasm Repository. All cases were reviewed by six experienced dermatopathologists and only cases with an unequivocal consensus diagnosis were included in the study. Spitz nevi were defined as benign melanocytic nevi composed of large epithelioid, oval, or spindled melanocytes arranged in nests and/or fascicles (Figures 1a–c). Spitzoid melanoma was defined as a melanoma, histopathologically resembling a Spitz nevus and composed of large epithelioid melanocytes. Histopathological criteria used in the evaluation and classification of a lesion as a Spitzoid melanoma included (but were not limited to) the following: asymmetry, sheet-like growth pattern, expansile nodular growth, lack of maturation, ulceration of the epidermis, presence of mitotic figures, deep or atypical mitotic figures, deep extension into the subcutis, or large size (>1 cm) (Figures 1d–f). The following characteristics were recorded for Spitzoid melanomas: tumor thickness, primary anatomic location, age, gender, presence or absence of ulceration, and mitotic rate in accordance with the American Joint Committee on Cancer (AJCC). In addition, 21 conventional melanocytic nevi were also included. Clinical and histopathological data were recorded for all cases. Two 1-mm cores were obtained from each sample for DNA extraction. Adjacent normal tissue was used as a control. Data for the conventional melanoma samples were reused from a prior study.7

(a) A Spitz nevus (G728T) presenting as a symmetric, wedge-shaped and well-circumscribed melanocytic proliferation. (b) Vertically oriented nests of melanocytes with clefts between them and the surrounding hyperplastic epidermis. (c) The melanocytes are monomorphous and mitotic figures are absent. (d) A Spitzoid melanoma (G762T) showing asymmetric growth pattern. (e) The melanocytic nests vary markedly in size and shape and focally form sheets. The arrow marks a narrow ulceration of the epidermis. (f) The melanocytes are pleomorphic and display prominent nucleoli. The two arrows point to mitotic figures in the dermis.
Exome Sequencing and Variant Calling
Exome sequencing and somatic variant calling was performed as described.3, 7 We used the NimbleGen SeqCap EZ Exome v.2 Capture Kit, and performed paired-end sequencing on Illumina HiSeq. We used bwa and SAMtools for read alignment and single-nucleotide variant calling, using fully validated filtering criteria for identifying high-quality DNA variants.3, 7 Tumor DNA sequencing was performed with a mean coverage of 231±56 independent reads per base, mean error rate of 0.44%, and with 96% of the targeted bases covered with at least 20 independent reads. Normal DNA samples were sequenced with a mean coverage of 125±26 independent reads per base, mean error rate of 0.36%, and with 93% of the targeted bases covered with at least 20 independent reads. Owing to the slightly higher error rate compared with our earlier sequencing effort in conventional melanoma, we manually reviewed and filtered the top recurrent somatic hits in our samples.
Sanger Validation
The mutations in PRKAR1A and IDH1 were validated by Sanger sequencing of the DNA extracted from the formalin-fixed and paraffin-embedded tumors using forward 5′-GAGTGCCAGCTTTACATGCC-3′ and reverse: 5′-CCGCATCTTCCTCCGTGTAG-3′ primers that amplified 211 bp in PRKAR1A, and 5'- GGCACGGTCTTCAGAGAAGCC-3' and 5'-TGCCAACATGACTTACTTGATCCC-3' primers that amplified 118 bp in IDH1, each surrounding the mutation site.
Copy Number Variant Calling
Sequenza12 was used to derive allele-specific copy number, ploidy, and tumor purity from our data. We developed a script to extract arm-specific losses and gains from the Sequenza segmentation data. The normalized fold depth ratio was visualized using Circos.13
Survival Analysis
We assessed genetic changes for their effect on survival, using the univariate Cox proportional hazard model. Three common statistical tests were performed, namely likelihood ratio test, Wald test, and score test. Common gene mutations and chromosomal aberrations were represented as Boolean values, that is, true and false, and measured across the following known melanoma driver genes and mutations: BRAF (position 600), NRAS (positions 12,13, and 61), and any predicted damaging mutations in NF1, TP53, CDKN2A, ARID2, PTEN, RAC1, CYP7B1, and PPP6C. Chromosomal aberrations were recorded for each chromosome arm separately.
Machine Learning and Sample Classification
Based on similar features as discussed above, a decision-tree learning approach was used to classify nevi and melanomas based on exome sequencing data. Additional features included the total number of somatic mutations and chromosomal aberrations assessed for common alternations found in melanoma, that is, 6q, 8p, 9p, and 10q losses, as well as, 1q, 6p, 7p, 7q, 8q, 17q, and 20q gains. A decision-tree classifier14 in the Scikit-learn package performed the decision-tree learning. The package allows the user to input tree complexity, that is, the number of decision node levels, and performs feature selection given a set of input features. The classifier was trained in two stages. In the first stage, we performed decision-tree learning using a leave-one-out cross-validation. We then increased tree complexity by one for each run. This stage provided the optimal tree complexity for performing the classification, as well as the mean F1 score measured across the folds (we use the term accuracy for this measure across the remainder of the manuscript). In a second stage, we ran decision-tree learning over the whole data set, setting the tree complexity to the one determined in the first stage. This stage produced a final tree with optimal features selected, and optimal cut-points for those features. We used all available patients for comparing Spitz nevi to Spitzoid melanomas. The algorithm in the Scikit-learn package performed weighted sampling to balance the number of samples in comparing conventional nevi to melanoma.
MC1R Single-Nucleotide Variant Status
MC1R germline variants were classified as functional ‘R’ or non-function ‘r’ alleles based on published data15, 16, 17 or, for novel variants, based on in silico predictions by PolyPhen and Sift. Unlike ‘r’ alleles, R variants are thought to be damaging to protein function and/or known to be associated with red hair and light skin phenotypes. The members of each allele group are shown in Supplementary Table 2.
20/20 Analysis
We classified the top mutated genes based on the 20/20 heuristic rule.7, 18 The rule selects genes in which 20% or more of the observed somatic mutations are at recurrent sites, or in which 20% or more of the observed somatic mutations are inactivating (ie, nonsense mutations, splice-site mutations, or InDels). Additionally, we used the Fisher’s exact test to compare the number of samples with somatic mutations in Spitzoid and conventional melanoma.
Results and discussion
Sample Cohort
The Spitz nevi cohort consisted of 29 patients, 15 females and 14 males (F:M=1.1:1) that ranged in age between 1 to 35 years old (mean 8.8; median 7) (see Supplementary Table 1). The lesions were distributed, in decreasing order, on the lower extremity (12) head and neck (7) upper extremity (7) and trunk (3). The follow-up ranged between 5 and 25 years with a mean of 14.6 years and a median of 14 years. All patients were alive and with no evidence of recurrence or metastasis at the conclusion of the study. The Spitzoid melanoma cohort consistent of patients 34 to 89 years old (mean 64; median 65). There was no gender predominance. The lesions were distributed as follows in a decreasing order: trunk (8) lower extremity (6) upper extremity (7) and head and neck (6). Tumor thickness ranged between 1 and 9 mm, with a mean of 3.7 mm and a median of 3.1 mm. The follow-up ranged between 0.5 and 15.5 years, with a mean of 6.6 years and a median of 7 years. Seventeen patients were alive with no evidence of recurrence or metastasis, 6 were dead of disease, 1 expired of unknown cause, 2 patients were alive with disease, and 1 patient was lost to follow-up at the conclusion of the study. The 21 conventional nevi came from patients between 3 months and 14 years of age (mean age=8.5 and median=11 years); 10 males and 11 females. In decreasing order, the location of the nevi was: head and neck (14) trunk (6) and lower extremity (1). The follow-up ranged between 3 and 12 years, with a mean of 9.9 and median of 11 years. The 133 conventional melanoma samples, all from sun-exposed skin, have been described earlier.7 They consisted of 105 metastatic, and 28 primary samples. The majority of the latter were nodular melanomas (12) followed by superficial spreading melanoma (6) desmoplastic melanoma (1) spindle cell melanoma (1) lentigo maligna melanoma (1) and 7 melanomas of unknown type. Tumor thickness ranged between 0.2 and 22 mm, with a mean of 3.9 mm and a median of 2.6 mm for the full cohort, and between 0.55 and 22 mm, with a mean of 3.9 mm and a median of 3.3 mm for the primary samples only.
Mutational Landscape
The number of somatic silent and non-silent single-nucleotide variants, splice-site variants, and InDels differed markedly among the samples (Figure 2,Table 1, and Supplementary Table 1). The number of mutations was significantly different between nevi and melanoma (both of the Spitzoid and conventional type), with 9±3 and 758±97 mutations in nevi and conventional melanomas (P=2.35E−12), respectively, and 34±9 and 747±138 mutations in Spitz nevi and Spitzoid melanomas (P=2.09E−05), respectively. On the other hand, there was no statistical difference in the mutational counts between the two types of nevi, and the two types of melanoma. All subtypes showed a characteristic UV mutation spectrum with ~90% C>T mutations in the dipyrimidine context (Figure 3).

Mutational landscape of Spitz nevi, Spitzoid melanomas compared with conventional nevi and melanomas. The 133 conventional melanomas correspond to paired samples from a previous publication.7

Mutation spectrum. Observed mutations in non-dipyrimidines (left) and dipyrimidines (right).
There was a large overlap and commonalities in driver genes in Spitzoid melanomas and melanoma.7 Particularly, mutations affecting the MAPK pathway were present in 66% of the lesions, with 37% (10/27) BRAF-mutant, 18.5% (5/27) RAS-mutant, and 11.1% (3/27) NF1-mutant Spitzoid melanomas. NF1-mutant Spitzoid melanomas harbored additional RASopathy gene mutations, as recently reported in conventional NF1-mutant melanomas.7 Of the three NF1-mutant Spitzoid melanomas, one sample harbored two RASA2 (Pro530Leu, Gln500Ter), one a PTPN11 (Thr73Ile), and another a MAP2K1 (Pro124Leu) mutation (Supplementary Table 3). Spitzoid melanomas featured additional hotspot or inactivating mutations typically found in melanoma, including changes in CDKN2A, TP53, RAC1, PTEN, IDH1, and ARID2 (Supplementary Table 3). Our data, therefore, support the notion that conventional melanoma and Spitzoid melanoma are genetically similar, with undistinguishable somatic mutation counts, typical ultraviolet signature, and the same set of driver genes. These findings thus shed light on the ongoing controversy about whether these two lesions should be regarded as distinct entities.19, 20 The data also do not support a prior assessment that Spitzoid melanomas share a low-ultraviolet origin with Spitz nevi.11 We would like to note that the current study did not assess the presence of kinase fusion in Spitz nevi and Spitzoid melanomas.10 We repeated above analyses, comparing the sequenced Spitzoid melanomas, all primaries, to data from primary conventional melanoma only (Supplementary Figure 1). As with the full cohort of conventional melanoma, we found no statistical difference in mutational counts, and large overlap in the known melanoma driver genes.
The majority (18/21) of conventional nevi harbored known oncogenes, with 47.6% (10/21) BRAF and 38% (8/21) NRAS mutations. In contrast, oncogenic HRAS p.Gly13/Gln61 mutations were present in only 17.2% (5/29) of Spitz nevi, as described.21, 22 A search for additional mutations across key melanoma drivers7 revealed that one conventional nevus had the IDH1Arg132Cys mutant gene (Figure 4). Taken together, our data show that nevi, the conventional and Spitz type, harbor few ultraviolet mutations, and, with the exception of BRAF, NRAS, and HRAS, lack additional changes in melanoma driver genes. Our finding that at least one nevus harbored IDH1Arg132Cys suggests a transition to a malignant state, as recently demonstrated in a large study showing progressive accumulation of mutations in the transition to malignant melanoma.23 Approximately 55% of Spitz nevi harbor kinase fusions,10 which was not assessed in the current study, and which likely account for the primary mutational event in the HRASWT lesions.

Chromatogram of PRKAR1AK92* in G762T Spitzoid melanoma (top); and IDH1R132C in 774T1 Spitz nevus and G744 Spitzoid melanoma (bottom). The amino-acid sequences adjacent to the mutation site is indicated.
Gene Mutation Burden
Our data allowed us to tabulate top mutated genes in our sequenced cohort. Given our small sample size, we used the 20/20 rule as organizing principle. The rule identifies presumed oncogenes and tumor suppressors by tabulating and thresholding, per gene, the ratio of recurrent and inactivating mutations, respectively. BRAF, NRAS, and HRAS were the only recurrent oncogenic signature in conventional and Spitz nevi, and there was no gene that displayed a tumor suppressor signature (Supplementary Tables 4a and b). On the other hand, Spitzoid melanomas showed many of the same top mutated genes as conventional melanomas (Tables 2 and 3), including a recurrent signature in BRAF and IDH1 and inactivating signature in ARID2 and TP53. Interestingly, the 20/20 rule identified MEN1 and PRKAR1A as potential tumor suppressors in Spitzoid melanomas as both genes harbored mutations leading to early termination (Table 3 and Figure 4). Germline inactivating mutations in PRKAR1A cause multiple neoplasia syndrome Carney complex.24 The gene encodes the regulatory subunit type I-α of cAMP-dependent kinase, and loss-of-function increases protein kinase A (PKA) activity leading to activated MAPK.25 Among other anomalies, Carney complex patients feature intense skin pigmentation, pigmented epithelioid melanocytomas,26 and pituitary adenomas.27 Pituitary adenomas are also a hallmark of MEN1.27 These mutations occur rarely in conventional melanoma, suggesting involvement of the PKA pathway in the genesis of Spitzoid melanomas.
Copy Number Variant Analysis
Conventional nevi did not show any region-wide copy number alterations (Figure 5). In Spitz nevi, we observed known 11p amplifications,22 in addition to hemizygous chromosome 9 deletions (Figure 5 and Supplementary Figure 2), also described earlier.28 In contrast, Spitzoid melanomas mirrored typical copy number changes observed in melanoma, including 6q, 8p, and 9p losses, as well as 6p and 8q gains (Figure 5).

Copy number alterations as shown by normalized log R ratio. These figures are plotted by Circos. (a) Spitz nevus; (b) Spitzoid melanoma; (c) conventional nevus; and (d) conventional melanoma. Blue indicates copy number losses, and red indicates copy number gains.
Comparative Genetic Analysis
Using a statistical comparison of gene-level mutation burden, we identified higher mutation rate in DNMT3A in Spitzoid melanomas compared with melanomas (Supplementary Table 5) with three predicted damaging DNMT3A mutations in Spitzoid melanoma. Recurrent DNMT3A mutations are associated with acute myeloid leukemia.29 Interestingly, an additional mutation was observed in Spitz nevi, supporting a possible role of DNA methylation in the pathogenesis of Spitzoid lesions.
We further used a machine learning approach to identify key differences among the sequenced lesions with regard to the identified driver mutations, and/or chromosomal aberrations. The number of somatic coding single-nucleotide variants (silent and non-silent) across the exome was selected as the major distinguishing feature between the benign and malignant conventional lesions. For our samples, this feature allows to distinguish between conventional melanoma and nevi with an accuracy of 93.5% (Supplementary Figure 3, right). Classification performance was 93.9% when comparing nevi with primary conventional melanoma only. For the comparison between Spitz nevus vs Spitzoid melanoma, an additional node counting the number of known melanoma driver mutations was essential to achieve a classification performance of 92.9% (Supplementary Figure 3, left). The below 100% accuracy is caused by some melanomas that do not exhibit key melanoma mutations or chromosomal aberrations. We also investigated whether data from small sequencing panels, rather than exome data, achieve similar classification performance. We thus simulated a targeted sequencing array covering 190 melanoma and other actionable cancer driver genes30 by filtering our exome data for mutations in those genes only. We assumed that chromosome arm-level copy number assessment is still feasible using the smaller panel, for example, by using dedicated copy number variant sequencing probes.31 As seen in Supplementary Figure 4, due to the smaller capture region, the total number of observed somatic mutations is a less reliable decision criterion for distinguishing conventional nevi from conventional melanoma, and the algorithm selects the number of observed copy number events as an additional decision node, achieving a classification accuracy of 92.8% (Supplementary Figure 4, right). The decision tree for Spitzoid lesions is unchanged to the one discussed above, showing a classification accuracy of 91.1% for the simulated targeted array (Supplementary Figure 4, left).
Survival Analysis
We analyzed whether any of the key mutational events in melanoma have an influence on patient survival. The tested mutations did not show a significant correlation with survival (Supplementary Table 6), and while some copy number alterations were associated with survival, none achieved significance after multiple hypothesis correction.
MC1R Analysis
Given the PRKAR1A findings that indicated possible involvement of the PKA pathway, we explored whether there were differences in MC1R variants among the skin lesions. Overall, there was no large difference among the subtypes, with 44.8%, 33.3%, 48.1%, and 44.4% of Spitz nevi, conventional nevi, Spitzoid melanoma, and conventional melanoma harboring the MC1R ‘R’ variant (see Supplementary Table 2). MC1R Arg151Cys and Arg160Trp were present most frequently across the melanoma and nevi subtypes. There was a significant decrease in Arg151Cys frequency in Spitzoid melanoma compared with conventional melanoma (3.7% vs 22.6%, P=0.0238), and a significant increase for Arg160Trp (25.9% vs 11.2%, P=0.0438).
Limitation of the Approach
While our sequencing approach follows good practices using a fully validated data analysis pipeline, we would like to note a couple of limitations. We noticed a slight increase in sequencing error rate (0.4% vs 0.2%) in the FFPE samples compared with our previous melanoma data, which were largely derived from fresh-frozen tissue. Additionally, we collected two 1-mm cores from each sample for DNA extraction, with adjacent normal tissue used as a control. As a result, for a minority of collected normal tissue, we observed slight contamination with tumor cells, affecting somatic mutation calling. Also, we needed to exclude 4% (3/80) of the initially sequenced samples because of inadequate DNA quality. Finally, it should be noted that the discussed classification performance is valid for the samples included in this study, which does not contain ambiguous cases, such as atypical Spitz tumors.
Conclusion
We present exome-wide unbiased genomic sequencing data on Spitz nevi and conventional nevi, two benign lesions of the melanocytic lineage, and compare the findings to Spitzoid and conventional melanoma. Our data suggests that Spitzoid and conventional melanoma are genetically similar, with possible involvement of the PKA and DNA methylation pathways in Spitzoid lesions. We also find a striking difference in the number of somatic single-nucleotide variants, InDels, and copy number changes between the benign and malignant lesions, a finding that may be exploitable as we move sequencing into clinical practice.
References
Gartner JJ, Davis S, Wei X et al, Comparative exome sequencing of metastatic lesions provides insights into the mutational progression of melanoma. BMC Genomics 2012; 13: 505.
Hodis E, Watson IR, Kryukov GV et al, A landscape of driver mutations in melanoma. Cell 2012; 150: 251–263.
Krauthammer M, Kong Y, Ha BH et al, Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012; 44: 1006–1014.
Martin M, Masshofer L, Temming P et al, Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet 2013; 45: 933–936.
Stark MS, Woods SL, Gartside MG et al, Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat Genet 2012; 44: 165–169.
Wei X, Walia V, Lin JC et al, Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet 2011; 43: 442–446.
Krauthammer M, Kong Y, Bacchiocchi A et al, Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet 2015; 47: 996–1002.
Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015; 161: 1681–1696.
Charbel C, Fontaine RH, Malouf GG et al, NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol 2014; 134: 1067–1074.
Wiesner T, He J, Yelensky R et al, Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014; 5: 3116.
Bastian BC . The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 2014; 9: 239–271.
Favero F, Joshi T, Marquard AM et al, Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol 2015; 26: 64–70.
Krzywinski M, Schein J, Birol I et al, Circos: an information aesthetic for comparative genomics. Genome Res 2009; 19: 1639–1645.
Breiman L . Random Forests. Mach Learn 2001; 45: 5–32.
Beaumont KA, Shekar SN, Newton RA et al, Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum Mol Genet 2007; 16: 2249–2260.
Raimondi S, Sera F, Gandini S et al, MC1R variants, melanoma and red hair color phenotype: a meta-analysis. Int J Cancer 2008; 122: 2753–2760.
Pasquali E, Garcia-Borron JC, Fargnoli MC et al, MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer 2015; 136: 618–631.
Vogelstein B, Papadopoulos N, Velculescu VE et al, Cancer genome landscapes. Science 2013; 339: 1546–1558.
Mones JM, Ackerman AB . ‘Atypical’ Spitz's nevus, ‘malignant’ Spitz's nevus, and ‘metastasizing’ Spitz's nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol 2004; 26: 310–333.
Cerroni L . Spitzoid tumors: a matter of perspective? Am J Dermatopathol 2004; 26: 1–3.
Sarin KY, Sun BK, Bangs CD et al, Activating HRAS mutation in agminated Spitz nevi arising in a nevus spilus. JAMA Dermatol 2013; 149: 1077–1081.
Bastian BC, LeBoit PE, Pinkel D . Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol 2000; 157: 967–972.
Shain AH, Yeh I, Kovalyshyn I et al, The genetic evolution of melanoma from precursor lesions. N Engl J Med 2015; 373: 1926–1936.
Stergiopoulos SG, Stratakis CA . Human tumors associated with Carney complex and germline PRKAR1A mutations: a protein kinase A disease!. FEBS Lett 2003; 546: 59–64.
Robinson-White A, Hundley TR, Shiferaw M et al, Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinasesERK1/2. Hum Mol Genet 2003; 12: 1475–1484.
Zembowicz A, Knoepp SM, Bei T et al, Loss of expression of protein kinase a regulatory subunit 1alpha in pigmented epithelioid melanocytoma but not in melanoma or other melanocytic lesions. Am J Surg Pathol 2007; 31: 1764–1775.
Lecoq AL, Kamenicky P, Guiochon-Mantel A et al, Genetic mutations in sporadic pituitary adenomas—what to screen for? Nat Rev Endocrinol 2015; 11: 43–54.
Gerami P, Scolyer RA, Xu X et al, Risk assessment for atypical spitzoid melanocytic neoplasms using FISH to identify chromosomal copy number aberrations. Am J Surg Pathol 2013; 37: 676–684.
Ley TJ, Ding L, Walter MJ et al, DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010; 363: 2424–2433.
Hovelson DH, McDaniel AS, Cani AK et al, Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia 2015; 17: 385–399.
Mason-Suares H, Kim W, Grimmett L et al, Density matters: comparison of array platforms for detection of copy-number variation and copy-neutral abnormalities. Genet Med 2013; 15: 706–712.
Acknowledgements
Research reported in this publication was supported by the Yale SPORE in Skin Cancer, funded by the National Cancer Institute, US National Institutes of Health, under award number 1 P50 CA121974 (to MK, NP, and RH) and Gilead Sciences (to RL and YB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Data Deposition Statement
Sequencing data deposited in dbGAP with accesssion number phs000933.
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Lazova, R., Pornputtapong, N., Halaban, R. et al. Spitz nevi and Spitzoid melanomas: exome sequencing and comparison with conventional melanocytic nevi and melanomas. Mod Pathol 30, 640–649 (2017). https://doi.org/10.1038/modpathol.2016.237
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.237
This article is cited by
-
New and evolving concepts of melanocytic nevi and melanocytomas
Modern Pathology (2020)
-
Spitz melanoma is a distinct subset of spitzoid melanoma
Modern Pathology (2020)
-
Melanocytic tumors with MAP3K8 fusions: report of 33 cases with morphological-genetic correlations
Modern Pathology (2020)
-
Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma
Nature Cancer (2020)
-
Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas
Nature Medicine (2019)
