
Abstract
Fibrous hamartoma of infancy is a rare soft tissue lesion of infants and young children with characteristic triphasic morphology, which typically occurs in the axilla and less commonly in other locations. We reviewed 145 cases of fibrous hamartoma of infancy from our consultation archives. Cases occurred in 106 males and 39 females (mean age—15 months; range—birth to 14 years), and involved both typical sites (eg, axilla/back/upper arm) (n=69) and unusual locations (n=76). Six were congenital. The tumors presented as subcutaneous masses and ranged from 0.4 to 17 cm (mean 3 cm). All displayed triphasic morphology, but varied widely in the relative percentages of fat, fibroblastic fascicles, and primitive mesenchyme. Hyalinized zones with cracking artifact, mimicking giant cell fibroblastoma, were present in a 44 (30%) of cases; however FISH for PDGFB gene rearrangement was negative in five tested cases. In addition to classical fibrous hamartoma of infancy, two lesions contained large sarcomatous-appearing foci with high cellularity, high nuclear grade, and brisk mitotic activity. One occurred in a 10-month-old female as a new mass in a congenital fibrous hamartoma of infancy; the other occurred as a leg mass in a 6-year-old male. ETV6 gene rearrangement was negative in the tumor from the 10-month-old female. Genomic microarray (OncoScan) showed normal molecular karyotype in eight tested cases, whereas the two tumors with sarcomatous features showed a hyperdiploid/near tetraploid molecular karyotype with copy neutral loss of heterozygosity of chromosomes 1p and 11p, and loss of 10p, chromosome 14, and a large portion of chromosome 22q (22q11.23q13.33), respectively. Follow-up (52 patients; range: 1–208 months, median: 8 months) showed only two local recurrences and no metastases. Extensive local disease in the 10-month-old female with sarcomatous-appearing fibrous hamartoma of infancy necessitated forequarter amputation. In summary, our study confirms the classic clinicopathologic features, including the triphasic morphologic appearance of most cases. In contrast to earlier studies, our series illustrates a broader histologic spectrum than previously appreciated, including its close resemblance to giant cell fibroblastoma in one quarter of cases and the rare presence of ‘sarcomatous’ areas, the latter providing evidence that these are complex neoplasms rather than hamartomas.
Similar content being viewed by others
Main
Fibrous hamartoma of infancy, originally described in 1956 by Reye as ‘subdermal fibromatous tumor of infancy’, is a rare, distinctive soft tissue neoplasm of infants and young children showing characteristic triphasic morphology with an unusual admixture of bland fibroblastic fascicles, mature adipose tissue, and nodules of primitive myxoid mesenchyme.1 Reye considered them to represent a reactive phenomenon, based on his belief that they showed a progressive and orderly maturation to adult fibrous tissue.1 In 1965, however, Enzinger reported his experience with 30 such cases and suggested the term ‘fibrous hamartoma of infancy’, noting that the clinical presentation in infants and young children, absence of preceding injury, and uniformly organoid appearance of the tumors seemed more in keeping with a hamartomatous process, rather than a reparative one.2 Roughly 200 cases of fibrous hamartoma of infancy have been reported, in the form of clinical case series3, 4, 5, 6, 7, 8, 9 and case reports.10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49
Owing to its rarity, however, there have been no recent large clinicopathologic studies of fibrous hamartoma of infancy. Our study, representing the cumulative experience from four institutions, was undertaken to define its histologic spectrum, molecular abnormalities, biologic behavior, and to evaluate the previous concept of a ‘hamartoma.’
Material and methods
This study was approved by the Institutional Review Boards at Mayo Clinic, Emory University Hospital, Boston Children’s Hospital, and Children’s Hospital of Los Angeles. All available routinely stained and immunohistochemistry slides for 164 cases previously diagnosed as ‘fibrous hamartoma of infancy’ were retrieved from our collective institutional and consultation archives and re-reviewed by two of the authors (AA-I and ALF). The number of slides per case available for re-review ranged from 1 to 30. On re-review, 19 cases were thought to have insufficient material or to more likely be other diagnoses (eg, lipofibromatosis, desmoid-type fibromatosis) and were excluded, leaving a final study population of 145 cases. Clinical information, including patient follow-up, was obtained either from our institutional medical records or (for consultation cases) from the submitting pathologists and clinicians. For each case, the relative percentage of fibroblastic fascicles, adipose tissue, and primitive mesenchyme was recorded, as was the presence or absence of hyalinized zones with cracking artifact resembling giant cell fibroblastoma. We also evaluated each case for chronic inflammatory cell aggregates, necrosis, mitotic figures per 10 high power field, and for any other unusual histologic features. A component was considered ‘predominant’ if it occupied equal or more than 45% of the entire lesion. Immunohistochemical results for keratins, S100 protein, smooth muscle actins, desmin, CD34, ALK-1 protein, and beta-catenin protein were recorded, noting expression patterns in the fibroblastic, adipocytic, and primitive mesenchymal zones.
Fluorescent in situ hybridization (FISH) for platelet-derived growth factor receptor-beta (PDGFB) rearrangement/amplification (N=5) or ETV6 (N=1) gene rearrangement was performed on formalin-fixed, paraffin-embedded tissue sections, using previously published methods.50, 51
For genomic microarray studies, genomic DNA was extracted from formalin-fixed, paraffin-embedded cut slides, and quantitated utilizing a Qubit 2.0 Fluorometer (Life Technologies/Thermo Fisher Scientific; USA) and standard methods. The isolated DNA was then processed with the OncoScan FFPE Assay Kit (Affymetrix; Santa Clara, CA, USA). The technical details of this molecular inversion probe (MIP) assay have been previously described.52, 53, 54 Briefly, a MIP targeting a unique SNP or base pair of interest anneals to isolated DNA and circularizes with its complementary nucleotide. Single-stranded material is destroyed and the circular probe is released, cleaved, inverted, and amplified using universal primers. The biotinylated oligonucleotide is hybridized overnight to a microarray. Two microarray chips are used per sample and the entire process takes ~48 h. Microarray results were then visualized using the Chromosome Analysis Suite (ChAS) software (Affymetrix; Santa Clara, CA, USA).
Results
Clinical Features
Table 1 summarizes the clinical findings. The tumors occurred in 106 males and 39 females (male: female ratio=2.7) with a mean age of 15 months (range—birth to 14 years). In 132 cases (91%), the patients were <2 years of age; six cases (4%) were congenital. The tumors ranged from 0.4 to 17 cm in size (mean 3 cm), and occurred in the subcutaneous tissues of the axilla (n=25; 17%), back (n=24; 16%), upper arm (n=20; 14%), scrotum (n=13; 9%), chest wall (n=12; 8%), thigh (n=9; 6%), neck (n=8; 5%), breast (n=5; 4%), forearm (n=5; 4%), abdominal wall (n=4; 3%), buttock (n=4; 3%), cheek (n=3; 2%), foot (n=3; 2%), shoulder (n=3; 2%), finger (n=2; 1%), scalp (n=2; 1%), flank (n=1; 1%), hip (n=1; 1%), and orbit (n=1; 1%). One patient presented with multiple nodules involving the lower back. Six patients presented with changes to the overlying skin, with discoloration, edema, hypertrichosis, and skin tethering. None of the patients were known to have clinical features suggestive of any syndrome.
Two cases contained areas resembling undifferentiated sarcoma (see ‘Results’ section). The first of these presented in a newborn girl as a massive congenital lesion of the left arm, chest wall, and back. Magnetic resonance imaging (MRI) showed a heterogeneous-appearing lesion with a large component of mature adipose tissue. A needle biopsy, done at age 9 days, was interpreted as showing a ‘hemangioendothelioma’, presumably representing sampling only of highly vascular areas within the tumor. Continued clinical concern prompted a second biopsy at age 2 months, which was interpreted as a fibrolipomatous tumor, possibly lipofibromatosis. Shortly thereafter, the tumor continued to grow, requiring eventual debulkment of the shoulder region. The excised specimen from the shoulder was interpreted by an expert soft tissue pathologist as showing an undifferentiated high-grade sarcoma. A similar lesion, thought to most likely be loco-regional spread secondary to the prior surgery, was excised 1 month later from the back. The patient then received chemotherapy with vincristine, actinomycin, and cyclophosphamide, with a slight decrease in the size of the masses. She was then referred to Mayo Clinic, where radical tumor resection including forequarter amputation was performed, owing to the extensive nature of the tumor and the non-functional status of the left arm. Tissue from this specimen showed only typical fibrous hamartoma of infancy, without residual sarcoma. She is currently alive and without disease, 42 months after initial presentation.
The second such patient was a 6-year-old boy who presented with a slowly growing, non-tender 5.5 cm mass below the knee. Additional clinical information was not available for this patient.
Follow-up information was available for 52 patients with a range of 0.5–208 months (median 8 months). Local recurrences were seen in 2 patients (1%); no metastases were noted.
Pathologic Features
Table 2 summarizes the morphological findings. The morphological and immunohistochemical features of typical fibrous hamartoma of infancy are illustrated in Figures 1a–f. Grossly, the lesions were described as poorly delineated, variably myxoid, and cotaining variable amounts of adipose tissue and gray to tan fibrous tissue. Histologically, all tumors displayed at least in part classical triphasic morphology, with haphazardly arranged, intersecting fascicles of cytologically bland fibroblastic/myofibroblastic cells, resembling those seen in fibromatosis, mature adipose tissue, and highly vascular, myxoid nodules of primitive-appearing spindled to stellate cells. Chronic inflammatory cell aggregates were commonly present. The relative proportion of fat, fibroblastic fascicles, and primitive mesenchyme varied from case to case, with approximately one-third of cases showing a more or less even distribution of all three elements, and the remaining cases being essentially evenly split between those consisting principally of fibroblastic zones and those consisting chiefly of fat. Only five cases showed a predominance of primitive myxoid mesenchymal elements. Mitotic figures were infrequent and necrosis was absent.


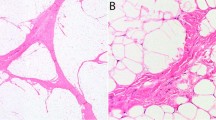
Typical triphasic morphology of fibrous hamartoma of infancy (a) with haphazardly arranged, intersecting fascicles of cytologically bland fibroblastic/myofibroblastic cells (b), mature adipose tissue (c), and highly vascular, myxoid nodules of primitive-appearing spindled to stellate cells (d,e). Smooth muscle actin expression, in a myofibroblastic pattern, was commonly seen in the fibroblastic zones (f). Areas mimicking giant cell fibroblastoma, characterized by abundant hyalinized collagen with cracking artifact, forming slit-like spaces lined by flattened to somewhat prominent tumor cells (g). Occasional multinucleated cells were present (h). These areas were strongly positive for CD34, further simulating giant cell fibroblastoma (i).
In 44 cases (30%) there were extensively hyalinized zones, consisting of broad mats of collagen with cracking artifact, forming slit-like spaces lined by flattened to somewhat prominent tumor cells (Figures 1g and h). These areas were typically strongly positive for CD34 (Figure 1i), and closely mimicked those found in giant cell fibroblastoma. For this reason, five such cases were tested by FISH for PDGFB rearrangement; all were negative.
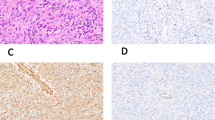
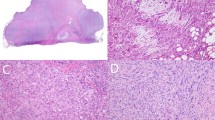
As noted above, two cases showed sarcomatous morphological features. The first of these, the congenital tumor in newborn female, showed an abrupt transition from areas of triphasic fibrous hamartoma of infancy to what appeared to be an undifferentiated-appearing spindled to round cell sarcoma, showing high cellularity, high nuclear grade, and very brisk mitotic activity (>10 mitotic figures/10 high powered fields) (Figures 2a–e). FISH for ETV6 rearrangement was performed twice on this case to exclude the unlikely possibility of infantile fibrosarcoma arising within fibrous hamartoma of infancy, with negative results. The second case, a leg mass from a 6-year-old boy, showed relative preservation of the organoid architecture seen in fibrous hamartoma of infancy as well as typical-appearing areas of banal fibroblastic, adipocytic, and primitive mesenchymal tissue. In addition, however, several nodules within this mass consisted of primitive-appearing round cells with high nuclear grade and mitotic activity, with production of an unusual, osteoid-like, densely eosinophilic matrix (Figures 3a–e).

Fibrous hamartoma of infancy with sarcomatous features, arising in the arm/axilla/back in a newborn girl. CT scan showed a large, heterogeneous lesion with a large adipocytic component (yellow arrow), within which arose a solid-appearing nodule (blue arrow) (a). The great majority of this very large mass consisted of typical triphasic fibrous hamartoma of infancy, shown at low (b), and higher power magnification (c). The typical fibrous hamartoma of infancy showed an abrupt transition (d) to a highly cellular, malignant-appearing spindle cell neoplasm with very high mitotic activity (e). Genomic microarray showed a hyperdiploid/near tetraploid molecular karyotype with gains of several chromosomes and copy neutral loss of heterozygosity of chromosomes 1p and 11p (f).


Fibrous hamartoma of infancy with sarcomatous features, arising in the leg from a 6-year-old boy. Much of the tumor consisted of typical triphasic fibrous hamartoma, seen at low (a) and higher (b) magnification. Within these areas of typical fibrous hamartoma multiple sarcomatous-appearing nodules were present, with overall preservation of the characteristic organoid architecture (c). The sarcomatous nodules were characterized by marked hypercellularity and malignant-appearing spindled and round cells with mitotic activity (d,e). In areas, the sarcomatous foci produced an unusual osteoid-like eosinophilic matrix (f). OncoScan array identified loss of 10p, chromosome 14, and large portion of chromosome 22q (22q11.23q13.33) (g).
Immunohistochemistry
By immunohistochemistry, 18 of 24 (75%) tested cases showed variable expression of smooth muscle actins in a myofibroblastic pattern, usually limited to fibroblastic areas, and occasionally in primitive mesenchyme. S100 protein was expressed by adipocytes in all 25 tested cases. CD34 expression was seen in the primitive mesenchymal component of all 21 tested cases, and was strongly positive in the giant cell fibroblastoma-like areas of 9 tested cases. Desmin was negative in 24 tested cases. A small number of tested cases were also negative for aberrant nuclear beta-catenin localization (N=3), keratins and/or epithelial membrane antigen (n=2), CD117 (n=1), and ALK-1 protein (n=1).
Genomic Microarray Findings
Details of the 10 tested cases are provided in Table 3. Briefly, testing by OncoScan showed a normal result in eight cases (six with classic triphasic histology, two with giant cell fibroblastoma-like features). The congenital fibrous hamartoma of infancy with sarcomatous features showed a hyperdiploid/near tetraploid molecular karyotype copy neutral loss of heterozygosity of chromosomes 1p and 11p (Figure 2f). In the malignant-appearing fibrous hamartoma of infancy from the 6-year-old, OncoScan identified loss of 10p, loss of chromosome 14, and loss of a portion of chromosome 22q (22q11.23q13.33) (Figure 3f).
Discussion
Fibrous hamartoma of infancy usually occurs in children <2 years of age, and presents as a congenital lesion in ~20% of cases.55 However, cases occurring in older children are well-documented.4, 8 The tumors are much more common in males, and most often present as a painless subcutaneous mass involving the axilla, trunk, upper arm, and external genitalia, although isolated cases of fibrous hamartoma of infancy have been reported in a wide variety of anatomical locations, including the scalp,16 foot,35 hand,24 and buttock.10 MRI has been reported to show an organized arrangement of fat with interspersed, heterogeneous soft tissue bands that may suggest this diagnosis in the appropriate clinical setting.41 Although isolated cases of fibrous hamartoma of infancy have been reported in patients with tuberous sclerosis22 and Williams syndrome,45 these appear to represent chance occurrences, rather than true syndromic association. The overwhelming majority of previously reported cases of fibrous hamartoma of infancy have been cured by simple excision, although ~15% have recurred locally in a non-destructive fashion,55 and rare cases have been reported to recur more than once.1, 2, 4 Clinically and/or histologically malignant fibrous hamartoma of infancy have not been reported.
The present series is the largest primary series of fibrous hamartoma of infancy to date, and includes the largest number of cases with clinical follow-up. We were only able to obtain clinical follow-up on 36% of our patients, a rate similar or better than that of previous large series of fibrous hamartoma of infancy, and likely reflecting the inherent difficulty in obtaining follow-up on children with ostensibly benign disease. The overall clinical features of the patients in the present series are similar to those previously reported, with a strong male predominance, a mean patient age of 15 months, and common involvement of the axilla, back, upper arm, chest wall, and external genital region. We found a somewhat lower rate of local recurrences than previously reported, likely due to greater efforts by pediatric surgeons in achieving negative resection margins. Our somewhat lower percentage of congenital lesions and the greater number of older patients likely reflect referral bias, as pathologists are presumably more likely to refer cases occurring in older patients.
Clearly the most unusual findings in the present study are the two fibrous hamartoma of infancy showing in part sarcomatous morphology. Although rare cases with atypical features, such as large size, rapid growth, infiltrative growth, and/or locally recurrence have been reported,2, 4, 9, 22, 23, 29, 35, 56 we are not aware of a previous report of an fibrous hamartoma of infancy showing overt sarcomatous features. The exceptional nature of these two cases prompted careful re-review of their morphology, which we believe contains incontrovertible areas of classical, benign-appearing, triphasic fibrous hamartoma of infancy (as illustrated in Figures 2b–d and 3a and b). Although the cellularity of the fibroblastic and primitive mesenchymal components of fibrous hamartoma of infancy varies somewhat from case to case, the morphological features of the sarcomatous foci in these two cases, with markedly elevated cellularity, high nuclear grade, and brisk mitotic activity far exceed that of ordinary fibrous hamartoma of infancy. Indeed, the initial biopsy from the patient with the congenital tumor was diagnosed as an ‘undifferentiated sarcoma’ by another expert soft tissue pathologist and was tested on two occasions for infantile fibrosarcoma-associated ETV6 rearrangements. The clinical significance of sarcomatous morphology in fibrous hamartoma of infancy remains, however, to be elucidated, especially as we were unable to obtain clinical follow-up on one of these two patients, despite intensive effort. The patient with the congenital, sarcomatous fibrous hamartoma of infancy, treated with radical tumor resection, is alive and well nearly 4 years after her initial presentation. Until we have a better collective understanding of the biological significance of these types of findings, we suggest the use of the term ‘fibrous hamartoma of infancy with sarcomatous morphology’ rather than ‘fibrous hamartoma of infancy with malignant progression’ to describe similar cases.
One important morphological feature of fibrous hamartoma of infancy that we believe warrants special attention are areas resembling another (potentially more aggressive) pediatric soft tissue tumor, giant cell fibroblastoma.57 Such areas are characterized by dense, hyalinized collagen, cracking artifact, and pseudoangiomatous, slit-like spaces lined by flattened and occasionally more prominent, CD34-positive fibroblastic cells. This feature may have been alluded to by Reye, who commented that some fibrous hamartoma of infancy contained ‘young fibrous tissue’ with wire-like collagen, resembling in his view areas that might be seen in plexiform neurofibroma.1 Enzinger drew greater attention to these areas, noting that ‘five of the lesions deviated markedly from the characteristic pattern by a distinctive fibrosing process which dominated the histologic picture’ and vaguely resembled a keloid.2 Fletcher et al4 referred to these areas as ‘disorderly fibrosis’ and speculated that they were a late stage in the maturation of fibrous hamartoma of infancy, an opinion echoed by Sotelo-Avila and Bale.9 The ‘pseudoangiomatous’ quality of these areas was first noted by Saab et al,8 who identified such areas in 53% of fibrous hamartoma of infancy, and showed them to be routinely CD34-positive. These authors commented that this ‘pseudoangiomatous pattern with disorderly fibrosis can simulate many other soft tissue tumors, including various types of hemangioma and other vascular tumors, neurofibroma, schwannoma, Gardner fibroma, giant cell fibroblastoma, and solitary fibrous tumor’.8 Similar CD34-positive pseudoangiomatous areas were present in 44 cases (30%) in our series, 5 of which closely mimicked giant cell fibroblastoma. However, these 5 cases were negative for PDGFB rearrangement/amplification, the molecular hallmark of giant cell fibroblastoma, and dermatofibrosarcoma protuberans,58, 59 by FISH, reinforcing their designation as fibrous hamartoma of infancy.
Relatively little is known about the genetic events, if any, that underly fibrous hamartoma of infancy. A small number of cases have been reported to show cytogenetic abnormalities, including one case with a complex karyotype including t(6;12;8)(q25;q24.3;q13), another with complex structural rearrangements involving chromosomes 1, 2, 4, and 17, and a third showing the reciprocal translocation t(2;3)(q31;q21).28, 36, 43 These genetic findings and the locally recurring potential of some tumors supports the theory that fibrous hamartoma of infancy is a neoplastic, rather than hamartomatous process. Our study identified genomic copy number changes by microarray in two of eight cases. Interestingly, both lesions showing such copy number changes also showed sarcomatous features microscopically. One case showed especially complex abnormalities with a hyperdiploid/near tetraploid copy state and structural alterations and copy neutral loss of heterozygosity. Such a complex molecular karyotype would be expected in malignant tumors and can often be seen in high-grade sarcomas. The other sarcomatous case however, showed fewer abnormalities, without any that would suggest behavior. Interestingly, none of the previously reported cytogenetic abnormalities were seen in our two sarcomatous cases. However, the OncoScan platform is not able to detect balanced structural rearangements that do not result in net loss or gain of genomic material, and thus the normal result in the remaining six cases does not exclude such abnormalities.
The differential diagnosis of fibrous hamartoma of infancy is broad, and dependent on the relative proportion of fat, mature fibrous tissue and primitive mesenchymal elements. Predominantly fatty tumors should be distinguished from lipofibromatosis, the ‘lipofibromatosis-like neural tumor’60 and maturing lipoblastoma. Lipofibromatosis usually involves the distal extremities, unlike fibrous hamartoma of infancy, and consists of an infiltrative admixture of mature fat, lipoblast-like cells and short fascicles of bland, fibroblastic cells.61 The ‘lipofibromatosis-like neural tumor’, very recently described by Agaram et al,60 is characterized by occurrence in the superficial soft tissues of very young children, an infiltrative growth pattern, variable cytological atypia, S100 protein and CD34 expression, and rearrangements of the NTRK1 gene. Lipoblastomas are better circumscribed than are fibrous hamartoma of infancy, with a distinctly lobular pattern of growth, and typically contain at least small myxoid foci with a well-developed capillary network and scattered lipoblasts.62 Rearrangements of the PLAG1 gene are seen in lipoblastomas, and are not known to occur in fibrous hamartoma of infancy.63 For chiefly fibroblastic fibrous hamartoma of infancy, the differential diagnosis includes desmoid-type fibromatosis, myofibroma, and calcifying aponeurotic fibroma. Desmoid-type fibromatoses are rare in very young children, and consist of a compact proliferation of ‘active-appearing’, bland myofibroblastic cells arranged in long, sweeping fascicles, surrounding a thin-walled, dilated vasculature. A significant percentage of desmoid-type fibromatoses show aberrant nuclear accumulation of beta-catenin protein64 and mutations of the CTNNB1 gene;65 beta-catenin nuclear accumulation is not seen in fibrous hamartoma of infancy.66 Myofibromas show a distinctive biphasic pattern of growth, with (generally) peripheral zones consisting of hyalinized and myoid-appearing spindled cells and more cellular primitive cells, sometimes with necrosis and/or calcification towards the center of the lesion. Calcifying aponeurotic fibromas contain distinctive calcified, vaguely chondroid-appearing nodules surrounded by epithelioid cells, often with associated osteoclast-like giant cells, in addition to infiltrative fascicles of fibroblastic cells.67 It has been recently shown that calcifying aponeurotic fibromas are characterized genetically by an FN1–EGF fusion gene.68 Fibrous hamartoma of infancy showing a predominance of the primitive mesenchymal component and/or prominent hyalinization with cracking artifact are most likely to be confused with giant cell fibroblastoma, a differential diagnosis discussed above. Finally, those exceptionally rare fibrous hamartoma of infancy that contain sarcomatous areas must be distinguished from other pediatric sarcomas, in particular infantile fibrosarcoma and spindle cell rhabdomyosarcoma. Ancillary testing for ETV6 rearrangements, characteristic of infantile fibrosarcoma69 and immunohistochemistry for desmin, myogenin and MyoD1 (expressed by spindle-cell rhabdomyosarcoma but not fibrous hamartoma of infancy) should facilitate these distinctions.
In summary, we have reported the clinicopathological, immunohistochemical, and genetic features of the largest series of fibrous hamartomas of infancy to date. Our results confirm and extend prior observations about this distinctive pediatric soft tissue tumor, in particular its potential to mimic another, more aggressive pediatric lesion, giant cell fibroblastoma. Exceptionally, fibrous hamartoma of infancy appears to have potential to progress morphologically and to show features of a primitive spindle cell or round cell sarcoma, as well as genetic changes (loss of heterozygosity) suggestive of malignancy. The clinical significance of this finding remains to be determined. This notwithstanding, the clinical and morphological features of fibrous hamartoma strongly suggest that it is a neoplastic process, rather than a hamartoma.
References
Reye RD . A consideration of certain subdermal fibromatous tumours of infancy. J Pathol Bacteriol 1956;72:149–154.
Enzinger FM . Fibrous hamartoma of infancy. Cancer 1965;18:241–248.
Carretto E, Dall'Igna P, Alaggio R et al. Fibrous hamartoma of infancy: an Italian multi-institutional experience. J Am Acad Dermatol 2006;54:800–803.
Fletcher CD, Powell G, van Noorden S et al. Fibrous hamartoma of infancy: a histochemical and immunohistochemical study. Histopathol 1988;12:65–74.
Michal M, Mukensnabl P, Chlumska A et al. Fibrous hamartoma of infancy. A study of eight cases with immunohistochemical and electron microscopical findings. Pathol Res Pract 1992;188:1049–1053.
Paller AS, Gonzalez-Crussi F, Sherman JO . Fibrous hamartoma of infancy. Eight additional cases and a review of the literature. Arch Dermatol 1989;125:88–91.
Popek EJ, Montgomery EA, Fourcroy JL . Fibrous hamartoma of infancy in the genital region: findings in 15 cases. J Urol 1994;152:990–993.
Saab ST, McClain CM, Coffin CM . Fibrous hamartoma of infancy: a clinicopathologic analysis of 60 cases. Am J Surg Pathol 2014;38:394–401.
Sotelo-Avila C, Bale PM . Subdermal fibrous hamartoma of infancy: pathology of 40 cases and differential diagnosis. Pediatr Pathol 1994;14:39–52.
Agrawal L, Bansal R, Singh J et al. Fibrous hamartoma of infancy in an unusual location—a case report. Gulf J Oncol 2010;52–54.
Albukerk J, Wexler H, Dana M et al. A case of fibrous hamartoma of infancy. J Pediatr Surg 1979;14:80–82.
Amer S . Fibrous hamartoma of infancy. J Coll Phys Surg Pak 2006;16:381–382.
Arioni C, Bellini C, Oddone M et al. Congenital fibrous hamartoma of the knee. Pediatr Radiol 2006;36:453–455.
Chang WC, Huang GS, Lee HS et al. Fibrous hamartoma of infancy at the wrist. Pediatr Int 2010;52:317–318.
Efem SE, Ekpo MD . Clinicopathological features of untreated fibrous hamartoma of infancy. J Clin Pathol 1993;46:522–524.
Eppley BL, Harruff R, Shah M et al. Fibrous hamartomas of the scalp in infancy. Plast Reconstr Surg 1994;94:195–197.
Greco MA, Schinella RA, Vuletin JC . Fibrous hamartoma of infancy: an ultrastructural study. Hum Pathol 1984;15:717–723.
Groisman G, Kerner H . A case of fibrous hamartoma of infancy in the scrotum including immunohistochemical findings. J Urol 1990;144:340–341.
Grynspan D, Meir K, Senger C et al. Cutaneous changes in fibrous hamartoma of infancy. J Cutan Pathol 2007;34:39–43.
Guo YK, Ning G, Zhao FM et al. Fibrous hamartoma of infancy mimicking teratoma in the parapharyngeal space on multidetector row CT. Pediatr Radiol 2011;41:785–787.
Gupta JC, Salgia KM, Sharma HS . Fibrous hamartoma of infancy: a case report. Indian J Med Sci 1966;20:507–508.
Han HJ, Lim GY, You CY . A large infiltrating fibrous hamartoma of infancy in the abdominal wall with rare associated tuberous sclerosis. Pediatr Radiol 2009;39:743–746.
Imaji R, Goto T, Takahashi Y et al. A case of recurrent and synchronous fibrous hamartoma of infancy. Pediatr Surg Int 2005;21:119–120.
Jebson PJ, Louis DS . Fibrous hamartoma of infancy in the hand: a case report. J Hand Surg Am 1997;22:740–742.
Jung PM, Hong EK . Fibrous hamartoma of infancy manifested as multiple nodules—a case report. J Korean Med Sci 1990;5:243–247.
Kang G, Suh YL, Han J et al. Fibrous hamartoma of infancy: an experience of a single institute. J Korean Surg Soc 2011;81:61–65.
Keskin M, Tosun Z, Toy H et al. Subdermal fibrous hamartoma of infancy in the hand. Scand J Plast Reconstr Surg Hand Surg 2007;41:211–214.
Lakshminarayanan R, Konia T, Welborn J . Fibrous hamartoma of infancy: a case report with associated cytogenetic findings. Arch Pathol Lab Med 2005;129:520–522.
McGowan Jt, Smith CD, Maize J Jr. et al. Giant fibrous hamartoma of infancy: a report of two cases and review of the literature. J Am Acad Dermatol 2011;64:579–586.
Melnick L, Berger EM, Elenitsas R et al. Fibrous hamartoma of infancy: a firm plaque presenting with hypertrichosis and hyperhidrosis. Pediatr Dermatol 2015;32:533–535.
Mitchell ML, di Sant'Agnese PA, Gerber JE . Fibrous hamartoma of infancy. Hum Pathol 1982;13:586–588.
Miyamoto M, Tsunoda R, Gembun Y et al. Recurrence of fibrous hamartoma of infancy excised 14 years after the primary surgery. J Neurosurg Pediatr 2010;5:136–139.
Monajemzadeh M, Vasei M, Kalantari M et al. Vulvar fibrous hamartoma of infancy: a rare case report and review of literature. J Low Genit Tract Dis 2013;17:92–94.
Morris SD, Robson A, Calonje E . Case 2: fibrous hamartoma of infancy. Clin Exp Dermatol 2002;27:533–534.
Robbins LB, Hoffman S, Kahn S . Fibrous hamartoma of infancy; case report. Plast Reconstr Surg 1970;46:197–200.
Rougemont AL, Fetni R, Murthy S et al. A complex translocation (6;12;8)(q25;q24.3;q13) in a fibrous hamartoma of infancy. Cancer Genet Cytogenet 2006;171:115–118.
Seelhammer T, Lilly K, Kaye VN et al. Asymptomatic nodule's on a child's back: Challenge. Fibrous hamartoma of infancy. Am J Dermatopathol 2010;32:98–99.
Seguier-Lipszyc E, Hermann G, Kaplinski C et al. Fibrous hamartoma of infancy. J Pediatr Surg 2011;46:753–755.
Sengar M, Mohta A, Manchanda V et al. Paratesticular fibrous hamartoma in an infant. Singapore Med J 2012;53:e63–e65.
Song YS, Lee IS, Kim HT et al. Fibrous hamartoma of infancy in the hand: unusual location and MR imaging findings. Skeletal Radiol 2010;39:1035–1038.
Stensby JD, Conces MR, Nacey NC . Benign fibrous hamartoma of infancy: a case of MR imaging paralleling histologic findings. Skeletal Radiol 2014;43:1639–1643.
Stock JA, Niku SD, Packer MG et al. Fibrous hamartoma of infancy: a report of two cases in the genital region. Urology 1995;45:130–131.
Tassano E, Nozza P, Tavella E et al. Cytogenetic characterization of a fibrous hamartoma of infancy with complex translocations. Cancer Genet Cytogenet 2010;201:66–69.
Thami GP, Jaswal R, Kanwar AJ . Fibrous hamartoma of infancy in the scrotum. Pediatr Dermatol 1998;15:326.
Togo T, Araki E, Ota M et al. Fibrous hamartoma of infancy in a patient with Williams syndrome. Br J Dermatol 2007;156:1052–1055.
Vecchio GM, Miano AE, Belfiore G et al. Fibrous hamartoma of infancy of the labium majus: a typical lesion in an unusual site. Pathologica 2010;102:71–74.
Vinayak RS, Kumar S, Chandana S et al. Fibrous hamartoma of infancy. Indian Dermatol Online J 2011;2:25–27.
Yoon TY, Kim JW . Fibrous hamartoma of infancy manifesting as multiple nodules with hypertrichosis. J Dermatol 2006;33:427–429.
Yu G, Wang Y, Wang G et al. Fibrous hamartoma of infancy: a clinical pathological analysis of seventeen cases. Int J Clin Exp Pathol 2015;8:3374–3377.
Abbott JJ, Erickson-Johnson M, Wang X et al. Gains of COL1A1-PDGFB genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Mod Pathol 2006;19:1512–1518.
Mathew S, Shurtleff SA, Raimondi SC . Novel cryptic, complex rearrangements involving ETV6-CBFA2 (TEL-AML1) genes identified by fluorescence in situ hybridization in pediatric patients with acute lymphoblastic leukemia. Genes, Chrom Cancer 2001;32:188–193.
Absalan F, Ronaghi M . Molecular inversion probe assay. Methods Mol Biol 2007;396:315–330.
Ji H, Welch K . Molecular inversion probe assay for allelic quantitation. Methods Mol Biol 2009;556:67–87.
Wang Y, Cottman M, Schiffman JD . Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet 2012;205:341–355.
Dickey GE, Sotelo-Avila C . Fibrous hamartoma of infancy: current review. Pediatr Dev Pathol 1999;2:236–243.
Ritchie EL, Gonzalez-Crussi F, Zaontz MR . Fibrous hamartoma of infancy masquerading as a rhabdomyosarcoma of the spermatic cord. J Urol 1988;140:800–801.
Shmookler BM, Enzinger FM, Weiss SW . Giant cell fibroblastoma. A juvenile form of dermatofibrosarcoma protuberans. Cancer 1989;64:2154–2161.
Macarenco RS, Zamolyi R, Franco MF et al. Genomic gains of COL1A1-PDFGB occur in the histologic evolution of giant cell fibroblastoma into dermatofibrosarcoma protuberans. Genes Chrom Cancer 2008;47:260–265.
Sirvent N, Maire G, Pedeutour F . Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chrom Cancer 2003;37:1–19.
Agaram NP, Zhang L, Sung YS et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol 2016;40:1407–1416.
Fetsch JF, Miettinen M, Laskin WB et al. A clinicopathologic study of 45 pediatric soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J Surg Pathol 2000;24:1491–1500.
Coffin CM . Lipoblastoma: an embryonal tumor of soft tissue related to organogenesis. Sem Diagn Pathol 1994;11:98–103.
Bartuma H, Domanski HA, Von Steyern FV et al. Cytogenetic and molecular cytogenetic findings in lipoblastoma. Cancer Genet Cytogenet 2008;183:60–63.
Bhattacharya B, Dilworth HP, Iacobuzio-Donahue C et al. Nuclear beta-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol 2005;29:653–659.
Le Guellec S, Soubeyran I, Rochaix P et al. CTNNB1 mutation analysis is a useful tool for the diagnosis of desmoid tumors: a study of 260 desmoid tumors and 191 potential morphologic mimics. Mod Pathol 2012;25:1551–1558.
Thway K, Gibson S, Ramsay A et al. Beta-catenin expression in pediatric fibroblastic and myofibroblastic lesions: a study of 100 cases. Ped Develop Pathol 2009;12:292–296.
Keasbey LE . Juvenile aponeurotic fibroma (calcifying fibroma); a distinctive tumor arising in the palms and soles of young children. Cancer 1953;6:338–346.
Puls F, Hofvander J, Magnusson L et al. FN1–EGF gene fusions are recurrent in calcifying aponeurotic fibroma. J Pathol 2016;238:502–507.
Adem C, Gisselsson D, Cin PD et al. ETV6 rearrangements in patients with infantile fibrosarcomas and congenital mesoblastic nephromas by fluorescence in situ hybridization. Mod Pathol 2001;14:1246–1251.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Al-Ibraheemi, A., Martinez, A., Weiss, S. et al. Fibrous hamartoma of infancy: a clinicopathologic study of 145 cases, including 2 with sarcomatous features. Mod Pathol 30, 474–485 (2017). https://doi.org/10.1038/modpathol.2016.215
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.215
This article is cited by
-
Fibrous hamartoma of infancy of the spinal cord resembling conus and filum, with a coexisting sacral dimple
Child's Nervous System (2024)
-
Test Yourself: 7-month-old female with an enlarging left axillary mass
Skeletal Radiology (2023)
-
Fibrous hamartoma of infancy: imaging findings
Pediatric Radiology (2023)
-
Lipoblastoma Arising in the Head and Neck: A Clinicopathologic Analysis of 20 Cases
Head and Neck Pathology (2023)
-
Giant dorsal lipofibromatosis in an infant: a case report
BMC Pediatrics (2022)
