
Abstract
Recurrent mutations in the myogenic transcription factor MYOD1 and PIK3CA were initially described in a subset of embryonal rhabdomyosarcomas. Recently, two independent studies demonstrated presence of MYODI (L122R) mutations as the basis to re-classify a spindle cell rhabdomyosarcoma, along with a sclerosing rhabdomyosarcoma, distinct from an embryonal rhabdomyosarcoma. We analyzed a much larger cohort of 49 primary rhabdomyosarcoma tumor samples of various subtypes, collected over a period of 9 years, for the presence of MYOD1 (L122R), PIK3CA (H1047), and PIK3CA (E542/E545) mutations, along with immunohistochemical analysis of desmin, myogenin, and MYOD1. Although activating PIK3CA mutations were absent across the sample set analyzed, we report 20% MYOD1 (L122R) mutation in rhabdomyosarcomas, found exclusively in 10 of 21 spindle cell and sclerosing rhabdomyosarcomas, occurring mostly in the head and neck region along with extremity sites (64%), than the paratesticular and intra-abdominal sites. Furthermore, while all 10 MYOD1 mutant spindle cell and sclerosing rhabdomyosarcoma samples showed diffuse and strong MYOD1 immunoexpression, 7 of 31 samples of rhabdomyosarcoma with wild-type MYOD1 were negative for MYOD1 expression. Clinically, a striking correlation was found between MYOD1 mutation and the clinical outcomes available for 15 of 21 cases: 5 of 7 patients with spindle cell and sclerosing rhabdomyosarcomas, harboring MYOD1 mutation, were alive-with-disease and 2 of 8 patients with spindle cell and sclerosing rhabdomyosarcomas, with mutant MYOD1, were free-of-disease. Taken together, we present the first report of MYOD1 (L122R) mutation in the largest cohort of 49 rhabdomyosarcomas reported so far, that are associated with a relatively aggressive clinical course. Moreover, consistent with the earlier two studies, this study further reinforces a relationship between spindle cell and the sclerosing rhabdomyosarcoma—now recognized as a single subtype, distinct from an embryonal rhabdomyosarcoma.
Similar content being viewed by others


Main
Rhabdomyosarcoma (RMS), a rare malignant tumor of skeletal muscle origin, is presently subtyped into an embryonal RMS, an alveolar RMS; a pleomorphic RMS and more recently, spindle cell and sclerosing RMS.1 Spindle cell RMS, initially considered as a variant of an embryonal RMS, is commonly identified in the paratesticular region of pediatric patients and is associated with a relatively better clinical outcome.2, 3 Subsequently, cases of spindle cell RMS were also described in adult patients, associated with a relatively more aggressive clinical course.4, 5, 6 Sclerosing RMS was identified as another distinctive variant of a RMS, histopathologically mimicking an extraskeletal osteosarcoma, an extraskeletal myxoid chondrosarcoma, a sclerosing epithelioid fibrosarcoma, and an angiosarcoma.7, 8, 9 Several studies have described a relationship between spindle cell and sclerosing RMSs that lately have emerged together as a distinct subtype of a RMS.4, 5, 10, 11, 12 Certain studies also demonstrated a relationship between some cases of embryonal RMS and spindle cell and sclerosing RMS.10, 12, 13
Various investigators have unraveled genetic events underlying cases of spindle cell and sclerosing RMS. Kohsaka et al14 initially described a recurrent MYOD1 (L122R) mutation defining a clinical subset of embryonal RMSs associated with PI3K-AKT pathway mutations. More recently, two independent studies demonstrated recurrent MYOD1 in adult spindle cell RMSs,15 and pediatric and adult sclerosing RMSs16 reinforcing a relationship between these two morphological variants of a RMS.
Here, we present a systematic analysis of activating mutations in MYOD1 and PIK3CA along with the expression of desmin, MYOD1, and myogenin in a much larger cohort of 49 primary samples of RMS collected over a period of 9 years: comprising of 21 spindle cell and sclerosing RMSs, 10 embryonal RMS,s 17 alveolar RMSs, and a single case of a pleomorphic RMS.
Materials and methods
Samples and Patients Description
The 49 cases of RMS included in the present study were selected, based on the availability of formalin-fixed and paraffin-embedded blocks, subsequent to a critical histopathologic review of 300 consecutive cases of RMS, diagnosed over a period of 9 years (2005–2013) at Tata Memorial Centre, Mumbai. Diagnostic criteria for various subtypes of RMS, including spindle cell/sclerosing RMS, were according to the recent World Health Organization classification of soft tissue tumors.1 Few cases of spindle cell RMS displayed focal areas of sclerosing RMS and vice versa. In such cases, the tumor was designated as spindle cell or sclerosing RMS, depending upon predominant tumor pattern present. All cases were reviewed by Bharat Rekhi, a pathologist, and the cases with an adequate amount of tumor content (more than 70%) in the corresponding paraffin blocks were included. Clinical and follow-up details were obtained with the help of electronic medical records, as well as telephonically.
Immunohistochemistry
Immunohistochemical staining was performed on formalin-fixed paraffin-embedded tissue sections by immunoperoxidase method using a MACH 2 Universal HRP-Polymer detection kit (Biocare, CA, USA), including 3′-3′-diaminobenzidine tetrahydrochloride as the chromogen. Appropriate positive and negative controls were included. The details of various antibody markers, including dilution and manufacturers, are enlisted in Supplementary Table 3. Immunohistochemical studies were performed on all cases.
Genomic DNA Extraction, PCR, and Sanger Sequencing
Genomic DNA from formalin-fixed and paraffin-embedded tissue blocks was isolated using QIAamp DNA formalin-fixed and paraffin-embedded tissue kit (Qiagen), as per manufacturer’s instructions. DNA concentration was determined by absorbance at 280 nm (NanoDrop 2000c, Thermo Scientific). Primers used for PCR amplifications of MYOD1 and PIK3CA amplicons are enlisted in Supplementary Table 4. PCR was performed in 25 μl reaction volume containing 50–100 ng of genomic DNA from formalin-fixed and paraffin-embedded blocks with 0.2 μM each of primer pairs using Veriti 96-Well Thermal Cycler (Applied Biosystems). For MYOD1 L122 and PIK3CA H1047 amplicons, PCR was carried out with initial hot-start denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, polymerization at 72 °C for 45 s, and final incubation for 10 min at 72 °C. For PIK3CA E542/E545 amplicon, PCR was carried out using all conditions as mentioned above with the annealing temperature of 60 °C for 30 s. PCR amplicons were purified using Nucleospin gel and PCR clean-up kit (Macherey-Nagel). Sequencing of purified PCR products was performed by Sanger sequencing and data were analyzed using Mutation Surveyor software V4.0.9.17
Statistical Analysis
Statistical analysis for various clinical features was carried out using IBM SPSS statistics software version 21 and significant differences were calculated using χ2 and Fisher’s exact test. Threshold for statistical significance was set at P≤0.05.
Results
Forty-nine cases of RMS in the present study included 17 cases of alveolar RMS, 10 of embryonal RMS, 21 of spindle cell and sclerosing RMS, and a single case of a pleomorphic RMS (Figure 1). Three out of 10 cases of alveolar RMS were tested for PAX-FOXO1 fusion transcript by RT-PCR and were found to be positive for PAX3-FOXO1 in two cases and PAX7-FOXO1 in a single case. Clinicopathologic features of 21 cases of spindle cell and sclerosing RMSs are enlisted in Table 1. There were 18 males and 3 females (6:1). Age range was 2–66 years. Mean age was 19.6 years and median age was 19 years. Site-wise, the tumors occurred in the head and neck region,11 paratesticular region,4 extremities,2 intra-abdominal region,3 and in the chest wall.1 Twenty-one cases of spindle cell and sclerosing RMS comprised 12 cases of spindle cell and 9 cases of sclerosing RMS.

Immunohistochemical and mutational analysis of 49 rhabdomyosarcoma (RMS) cases. Schematic representation of subtypes of RMS and overview of immunohistochemistry of desmin, myogenin and MyoD1 and MYOD1 (L122R), and PIK3CA (E542/E545 and H1047) mutation analysis. Black fill denotes sample harboring MYOD1 mutation; gray denotes wild-type MYOD1; and white denotes for data not available.
Histopathologic and Immunohistochemical Characteristics of the Primary Rhabdomyosarcomas
Histopathologically, cases of sclerosing RMS revealed pseudovascular-, nesting-, focal alveolar-, and cord-like arrangement of round to oval to short spindle-shaped tumor cells in a prominent hyaline or pseudochondroid stroma. Despite pseudoalveolar pattern noted in some cases of sclerosing RMS, none of those tumors displayed 'wreath-like' giant cells (Figure 2). Cases of spindle cell RMS displayed tumor cells with spindle-shaped nuclei, mostly arranged in long intersecting fascicles and occasionally in whorling patterns (Figure 3). Four cases of sclerosing RMS also displayed focal areas of tumor with spindle-shaped tumor cells (Figure 4). Certain cases of spindle cell and sclerosing RMS also displayed features of embryonal RMS, in the form of rhabdomyoblasts, especially cases of spindle cell RMS in tumors occurring in the paratesticular region. By immunohistochemistry, tumor cells in 21 of 21 cases were positive for desmin; in 18 of 20 were positive for MYOD1 in a diffuse pattern; in 17 of 19 were positive for myogenin (Figure 1); and in 6 of 11 cases were positive for smooth muscle actin, mostly in a focal pattern. In all cases, the tumor cells were positive for at least a single skeletal muscle-specific marker, namely MYOD1 and or myogenin. In a remaining single case, wherein MYOD1 was negative and myogenin was not performed, tumor cells were positive for myoglobin.

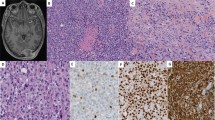
Immunohistochemical and mutational analysis of sclerosing rhabdomyosarcomas. Representative images of immunohistochemical analysis of MYOD1, myogenin, and MYOD1 (L122R) mutation in sclerosing rhabdomyosarcomas. Upper panel: Case 1. Sclerosing rhabdomyosarcoma (RMS) (I–III). Round to oval cells in a pseudovascular- and cord-like patterns in a dense hyalinized stroma. Hematoxylin and Eosin (H and E), x200. II. Diffuse MYOD1-positive immunostaining. Diaminobenzidine, x400. III. Focal myogenin-positive immunostaining. Diaminobenzidine, x400. Lower panel: Sanger sequencing chromatogram of MYOD1 (L122R, T>G or T>TG) is shown with reference sequences and mutation showing forward and reverse sequencing reads. Arrow indicates nucleotide position that harbors mutation.

Immunohistochemical and mutational analysis of spindle cell rhabdomyosarcomas. Representative images of immunohistochemical analysis of MYOD1, myogenin, and MYOD1 (L122R) mutation in spindle cell rhabdomyosarcomas. Upper panel: Case10. Spindle cell rhabdomyosarcoma (I–III). I. Spindle-shaped sarcomatous cells arranged in long intersecting fascicles. Hematoxylin and Eosin (H and E) x200. II. Diffuse MYOD1-positive immunostaining. Diaminobenzidine, x400. III. Myogenin-positive immunostaining. Diaminobenzidine, x400. Lower panel: Sanger sequencing chromatogram of MYOD1 (L122R, T>G or T>TG) is shown with reference sequences and mutation showing forward and reverse sequencing reads. Arrow indicates nucleotide position that harbors mutation.

Immunohistochemical and mutational analysis of a sclerosing rhabdomyosarcoma with focal areas of spindle cell rhabdomyosarcoma. Representative images of immunohistochemical analysis of MYOD1, myogenin, and MYOD1 (L122R) mutation in sclerosing rhabdomyosarcoma with focal spindle cell rhabdomyosarcomas. Case 2. Sclerosing rhabdomyosarcoma with focal spindle cells (I–III). I. Oval- to spindle-shaped cells in a dense hyalinized stroma. Hematoxylin and Eosin (H and E) x400. II. Diffuse MYOD1-positive immunostaining. Diaminobenzidine, x400. III. Focal myogenin-positive immunostaining. Diaminobenzidine, x400. Lower panel: Sanger sequencing chromatogram of MYOD1 (L122R, T>G or T>TG) is shown with reference sequences and mutation showing forward and reverse sequencing reads. Arrow indicates nucleotide position that harbors mutation.
Molecular Profiling for MYOD1 and PIK3CA Mutations across 49 Rhabdomyosarcomas
MYOD1 (L122R) mutation was found in 10 of 49 cases of RMS (Figure 1). Age-wise, 8 out of 10 cases were adult patients (>18 years of age) and 2 were pediatric patients (≤18 years of age) (P=0.08). Site-wise, 7 out of 10 cases revealing MYOD1 (L122R) mutation occurred in the head and neck region, whereas 2 cases occurred in the extremities and a single case occurred in the chest wall. (Table 1). Out of these 10 cases displaying MYOD1 mutation, 8 showed heterozygous mutations and 2 cases showed homozygous mutations. (Supplementary Figure 1 and Supplementary Table 1). All 10 cases displaying MYOD1 (L122R) mutation were of spindle cell and sclerosing RMS. None of the remaining 10 cases of embryonal RMS and 17 cases of alveolar RMS and the single case of pleomorphic RMS displayed MYOD1 (L122R) mutation. None of the 49 cases displayed PIK3CA (E542/E545) and PIK3CA (H1047) mutations (Figure 1). Among 21 cases of spindle cell and sclerosing RMS, more cases of sclerosing RMS (7/9) (78%) revealed MYOD1 mutations, as compared with spindle cell RMS (3/12) (25%) (P=0.03). Among seven cases of sclerosing RMS displaying MYOD1 mutations, four cases also displayed focal areas of tumor with spindle-shaped tumor cells. The remaining 11 cases of spindle cell and sclerosing RMS that showed wild-type MYOD1 included 9 cases of spindle cell RMS and 2 cases of sclerosing RMS. Seven of these 9 cases of spindle cell RMS occurred in abdominal sites, including 3 cases in the retroperitoneum and mesentery and 4 in the paratesticular region. The remaining two cases occurred in the head and neck region including face and soft palate. The two cases of sclerosing RMS, revealing wild-type MYOD1 occurred in the head and neck region (parotid and orbit) (Table 1).
Clinical Outcomes and their Correlation with Age and MYOD1 Mutational Status in Spindle Cell and Sclerosing Rhabdomyosarcoma
Treatment details were available in 19/21(90%) cases of spindle cell and sclerosing RMS. All the patients underwent surgical resections with adjuvant chemotherapy and radiotherapy in 9 cases; adjuvant chemotherapy (7 cases) and adjuvant radiotherapy (3 cases). Follow-up details (more than or equal to 6 months) were available in 15/21 cases (71%), over a period of 6–50 months (average=22 months, median=23 months). Overall, seven patients were alive-with-disease (over a duration of 6–33 months) and eight patients were free-of-disease (6–50 months) during the last follow up at our hospital. On comparing clinical outcomes between pediatric and adult patients, more number of adult patients was alive with disease (6/7) (86%), as compared with pediatric patients (1/7) (14%). Of the seven patients alive with disease, five were positive for MYOD1 mutation (71%), whereas two patients lacked MYOD1 mutation (29%). On the other hand, of eight patients free of disease, six were positive for wild-type MYOD1 (75%), whereas two patients were positive for MYOD1 mutation (25%). However, no statistically significant correlation (P=0.13) was observed between alive with disease and free of disease patients, with regard to MYOD1 mutation status (Table 1). Of these two cases, one presented with pulmonary metastasis, and other with pulmonary and lymph node metastasis (Supplementary Tables 1 and 2).
Discussion
The present study describes clinicopathologic features and analysis of MYOD1 (L122R) and PIK3CA (E542/E545, H1047) mutation profiling in 21 cases of spindle cell and sclerosing RMS, including 12 adult and 9 pediatric patients. These mutations were also tested in 17 cases of alveolar RMS; 10 cases of embryonal RMS; and in a single case of pleomorphic RMS.
Histopathologically, there were 12 cases of spindle cell RMS and 9 of sclerosing RMS. Four cases of sclerosing RMS also displayed focal areas of tumor with spindle cells, as noted earlier.8, 10, 11, 12 Whereas fascicular pattern was the most commonly observed tumor pattern in cases of spindle cell RMS, pseudovascular pattern was most commonly observed in cases of sclerosing RMS.4, 5, 7, 8, 9, 10, 11 By immunohistochemistry, all 21 cases displayed desmin positivity, along with MyoD1 positivity in 90% cases and myogenin positivity in 90% cases. Noteworthy, MyoD1 immunoexpression was diffuse in most cases of sclerosing RMS, as previously observed.10
While none of the cases of embryonal RMS, alveolar RMS, or pleomorphic RMS revealed MYOD1 mutation, consistent with the earlier reports;18 10 of 21 (48%) cases of spindle cell and sclerosing RMS were found to harbor MYOD1 (L122R) mutation, consistent with the earlier two reports wherein Szuhai et al15 reported the same in 41% cases of spindle cell RMS and Agaram et al16 reported this mutation in 56% cases of spindle cell and sclerosing RMS. Earlier, Kohsaka et al14 demonstrated MYOD1 (L122R) mutation in 10% cases of embryonal RMS.
Unlike the results from a study by Szuhai et al,15 wherein all seven the cases displayed homozygous mutation of MYOD1 (L122R), our study, similar to that of Agaram et al16 revealed cases of spindle cell and sclerosing RMS with heterozygous, as well as homozygous MYOD1 L122R mutations. Furthermore, similar to the latter study, we observed MYOD1 (L122R) mutation significantly more in the cases of sclerosing than spindle cell RMS (P=0.03).16 However, unlike Agaram et al,16 who observed MYOD1 mutation in all five cases of sclerosing RMS and in 36% cases of spindle cell RMS, we observed the same in 78% cases of sclerosing RMS and in 25% cases of spindle cell RMS. We believe the possible reason for the lower mutation frequency in cases of spindle cell RMS in the present study might be because there were four cases of paratesticular and three cases of abdominal spindle cell RMS, all that are generally not associated with MYOD1 (L122R) mutation, as recently described.19 A higher rate of MYOD1 mutation in cases of spindle cell RMS, reported in an earlier study, is attributable toward the fact most of those tumors from extremity sites, rather than intra-abdominal and paratesticular sites.15 Thus, the present study showed association of MYOD1 mutation, more with cases of spindle cell and sclerosing RMSs occurring in the head and neck and extremity sites, in contrast to those, especially with spindle cell morphology, occurring in the paratesticular and intra-abdominal sites, as noted in the two recent studies.16, 19
Therapeutically, most cases in the present study were treated with surgery and adjuvant chemotherapy with/without radiotherapy. On comparing clinical outcomes between adult and pediatric patients, more number of cases alive-with-disease was identified in the former group versus the latter. Though, not statistically significant, this suggests a trend for the spindle cell and sclerosing RMSs in adult patients to be relatively more clinically aggressive (P=0.08). This was likely as a result of 4/9 pediatric tumors in the present study occurring in paratesticular and intra-abdominal locations, wherein these tumors are associated with relatively better clinical outcomes.19 Furthermore, on comparing clinical outcomes in the cases showing MYOD1 mutation versus those lacking this mutation, we observed that a higher number of cases (5/7) (71%) with disease during their last follow up, in the form of recurrences and or metastasis were observed in MYOD1 mutant cases, although the difference was statistically not significant (P=0.34). Among the cases free-of-disease, there were less number of MYOD1 mutant cases (2/8) (25%) than those lacking this mutation (6/8) (75%).
On comparing clinical outcomes between MYOD1 mutant adult versus pediatric cases, more cases (5/6) (83%) alive-with-disease were seen in adult than in pediatric patients (1/6) (17%). Furthermore, more number of MYOD1 mutant cases, alive-with-disease were seen in adult than pediatric patients, indicating a relatively more aggressive clinical course in the former than the latter group. Contrastingly, Agaram et al16 observed a relatively more aggressive clinical course of MYOD1 mutant cases of spindle cell and sclerosing RMS in pediatric than in adult cases.16 This was in view of all such cases in their study group occurring sites other than paratesticular or abdominal region. Recently, Alaggio et al19 identified novel and recurrent VGLL2-related fusions in certain cases of infantile spindle cell RMS. Previously, Mosquera et al20 demonstrated recurrent NCOA2 gene rearrangement in cases of congenital/infantile spindle cell RMS. The same authors observed a relationship between NCOA2-rearranged spindle cell RMS occurring in young childhood and the so-called congenital RMS, which are associated with rearrangements at 8q13 locus (NCOA2), suggesting spindle cell RMSs as a heterogeneous group of tumors.
Furthermore, a small subset of cases of sclerosing RMS is known to be associated with PIK3CA mutations. Shukla et al21 reported the presence of PIK3CA mutation in 5% cases (3/60) of embryonal RMS, two of which were re-classified as sclerosing RMS. Although varying frequency of activating somatic mutations across different populations is known in the literature for EGFR,22 KRAS,23 BRAF,24 and PIK3CA25, 26 in lung, colorectal, and other cancers, absence of PIK3CA mutations in Indian cases of RMS is surprising. Of note, given low sample size of 49 tumors, the current study might not be adequately statistically powered to detect its occurrence if it exists at an altered frequency lower than 2–3%. However, to explore the therapeutic implication of PI3 kinase inhibitors in cases of RMS of Indian ethnicity, alterations in PTEN and AKT affecting alternate or redundant mechanism of PI3 kinase pathway activation need to be further analyzed.27, 28
In summary, the presence of specific MYOD1 (L122R) mutation in significant number of cases of spindle cell and sclerosing RMS, as noted in the present study, further reinforces a relationship between these two histopathological subtypes of RMS, now recognized as a single subtype of a RMS. Absence of this mutation in cases of embryonal RMS, RMS, and pleomorphic RMS indicates lack of the genetic relationship between spindle cell and sclerosing RMS with embryonal RMS that was previously considered, despite overlapping histopathologic and immunohistochemical features in some cases. MYOD1 immunostaining is frequently diffuse in cases of sclerosing RMS and constitutes as a useful immunohistochemical marker in these cases. MYOD1 (L122R) mutation was found to be more frequent in case of sclerosing RMS (irrespective of pediatric or adult patients), than in the spindle cell RMS, that constitutes as a relatively more heterogeneous group. Cases of spindle cell RMS occurring in the head and neck and extremity locations, in adult patients have higher frequency of MYOD1 mutation that those occurring in the paratesticular or intra-abdominal sites. PIK3CA mutations are rare in cases of spindle cell and sclerosing RMS and were not identified in any of our cases. Identification of specific MYOD1 (L122R) mutations in cases of sclerosing/spindle cell RMS probably indicates a more intensive treatment, as these cases are associated with a relatively aggressive clinical course. Thus genotyping for MYOD1 (L122R) mutation may preclude the requirement to perform immunohistochemical analysis in order to identify an aggressive subset of spindle cell and sclerosing RMSs, early on to help inform adoption of appropriate therapeutic regimen. At the same time, this discovery also seems to provide a direction toward avenues for targeted therapy in these cases.
References
Fletcher CDM Spindle cell/sclerosing rhabdomyosarcoma World Health Organization (WHO) classification of tumours of soft tissue and bone. Vol 5. Fourth Edn. IARC press: France, Lyon, 2013, 468 pp.
Cavazzana AO, Schmidt D, Ninfo V et al. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol 1992;16:229–235.
Leuschner I, Newton WA Jr., Schmidt D et al. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol 1993;17:221–230.
Mentzel T, Kuhnen C . Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch 2006;449:554–560.
Nascimento AF, Fletcher CD . Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol 2005;29:1106–1113.
Rubin BP, Hasserjian RP, Singer S et al. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol 1998;22:459–464.
Folpe AL, McKenney JK, Bridge JA et al. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol 2002;26:1175–1183.
Mentzel T, Katenkamp D . Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch 2000;436:305–311.
Wang J, Tu X, Sheng W . Sclerosing rhabdomyosarcoma: a clinicopathologic and immunohistochemical study of five cases. Am J Clin Pathol 2008;129:410–415.
Rekhi B, Singhvi T . Histopathological, immunohistochemical and molecular cytogenetic analysis of 21 spindle cell/sclerosing rhabdomyosarcomas. APMIS 2014;122:1144–1152.
Zhu L, Wang J . Sclerosing rhabdomyosarcoma: a clinicopathologic study of four cases with review of literature. Zhonghua Bing Li Xue Za Zhi 2007;36:587–591.
Chiles MC, Parham DM, Qualman SJ et al. Sclerosing rhabdomyosarcomas in children and adolescents: a clinicopathologic review of 13 cases from the Intergroup Rhabdomyosarcoma Study Group and Children's Oncology Group. Pediatr Dev Pathol 2004;7:583–594.
Croes R, Debiec-Rychter M, Cokelaere K et al. Adult sclerosing rhabdomyosarcoma: cytogenetic link with embryonal rhabdomyosarcoma. Virchows Arch 2005;446:64–67.
Kohsaka S, Shukla N, Ameur N et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 2014;46:595–600.
Szuhai K, de Jong D, Leung WY et al. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol 2014;232:300–307.
Agaram NP, Chen CL, Zhang L et al. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer 2014;53:779–787.
Minton JA, Flanagan SE, Ellard S . Mutation surveyor: software for DNA sequence analysis. Methods Mol Biol 2011;688:143–153.
Anand G, Shapiro DN, Dickman PS et al. Rhabdomyosarcomas do not contain mutations in the DNA binding domains of myogenic transcription factors. J Clin Invest 1994;93:5–9.
Alaggio R, Zhang L, Sung YS et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol 2016;40:224–235.
Mosquera JM, Sboner A, Zhang L et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 2013;52:538–550.
Shukla N, Ameur N, Yilmaz I et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res 2012;18:748–757.
Chougule A, Prabhash K, Noronha V et al. Frequency of EGFR mutations in 907 lung adenocarcioma patients of Indian ethnicity. PLoS One 2013;8:e76164.
Choughule A, Sharma R, Trivedi V et al. Coexistence of KRAS mutation with mutant but not wild-type EGFR predicts response to tyrosine-kinase inhibitors in human lung cancer. Br J Cancer 2014;111:2203–2204.
Rozek LS, Herron CM, Greenson JK et al. Smoking, gender, and ethnicity predict somatic BRAF mutations in colorectal cancer. Cancer Epidemiol Biomarkers Prev 2010;19:838–843.
Chong ML, Loh M, Thakkar B et al. Phosphatidylinositol-3-kinase pathway aberrations in gastric and colorectal cancer: meta-analysis, co-occurrence and ethnic variation. Int J Cancer 2014;134:1232–1238.
Zhang J, Zheng J, Yang Y et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: analysis of 1,110 cases. Sci Rep 2015;5:18678.
Yuan TL, Cantley LC . PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008;27:5497–5510.
Dutt A, Salvesen HB, Greulich H et al. Somatic mutations are present in all members of the AKT family in endometrial carcinoma. Br J Cancer 2009;101:1218–1219; author reply 20–1.
Acknowledgements
We would like to acknowledge Dr Chhavi Gupta, Resident, Department of Surgical Pathology, Tata Memorial Hospital, Mumbai, who helped in retrieving paraffin blocks of various cases, and all members of the Dutt laboratory for critically reading the manuscript. AD is supported by an Intermediate Fellowship from the Wellcome Trust/DBT India Alliance (IA/I/11/2500278), by a grant from DBT (BT/PR2372/AGR/36/696/2011) and intramural grants (Seed-In-Air 2897, TMH Plan Project 2712 and IRB 92). PU is supported by a senior research fellowship from CSIR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The support from immunohistochemistry laboratory, Department of Surgical pathology, and Tata Memorial Hospital is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Rekhi, B., Upadhyay, P., Ramteke, M. et al. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod Pathol 29, 1532–1540 (2016). https://doi.org/10.1038/modpathol.2016.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.144
This article is cited by
-
Biphenotypic Sinonasal Sarcoma with a Novel PAX7::PPARGC1 Fusion: Expanding the Spectrum of Gene Fusions Beyond the PAX3 Gene
Head and Neck Pathology (2023)
-
First Reported Case of Malignant Ectomesenchymoma with p.Leu122Arg Mutation in MYOD1 Gene: Extensive Intra- and Extracranial Tumor in a 15-Year-Old Female
Head and Neck Pathology (2023)
-
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Soft Tissue Tumors
Head and Neck Pathology (2022)
-
Neural stemness contributes to cell tumorigenicity
Cell & Bioscience (2021)
-
Methylation profiling reveals novel molecular classes of rhabdomyosarcoma
Scientific Reports (2021)

{kind=link}