
Abstract
Recent studies have demonstrated considerable genomic heterogeneity in both primary and metastatic renal cell carcinoma (RCC). This mutational diversity has serious implications for the development and implementation of targeted molecular therapies. We evaluated 39 cases of primary RCC tumors with their matched metastatic tumors to determine if the hallmark chromosomal anomalies of these tumors are preserved over the course of disease progression. Thirty-nine matched pairs of primary and metastatic RCCs (20 clear cell RCC, 16 papillary RCC, and 3 chromophobe RCC) were analyzed. All clear cell RCC and papillary RCC tumors were evaluated for chromosome 3p deletion, trisomy 7 and 17 using fluorescence in situ hybridization. Chromophobe RCC tumors were evaluated for genetic alterations in chromosomes 1, 2, 6, 10, and 17. Of the 20 clear cell RCC tumors, 18 primary tumors (90%) showed a deletion of chromosome 3p and were disomic for chromosomes 7 and 17. All molecular aberrations were conserved within the matched metastatic tumor. Of the 16 papillary RCC tumors, 10 primary tumors (62%) showed trisomy for both chromosomes 7 and 17 without 3p deletion. These molecular aberrations and others were conserved in the paired metastatic tumors. Of the three chromophobe RCC tumors, multiple genetic anomalies were identified in chromosomes 1, 2, 6, 10, and 17. These chromosomal aberrations were conserved in the matched metastatic tumors. Our results demonstrated genomic fidelity among the primary and metastatic lesions in RCCs. These findings may have important clinical and diagnostic implications.
Similar content being viewed by others

Main
Renal cell carcinoma (RCC) is a genetically heterogeneous disease with a wide spectrum of genetic and chromosomal aberrations, initially identified in the neoplastic tissues of both familial and sporadic clear cell and papillary RCC decades ago.1, 2, 3, 4 This mutational diversity currently has profound implications for treatment of metastatic RCC using targeted molecular therapies. Since the introduction of targeted therapies for the treatment of metastatic RCC, an understanding of genetic events occurring from the primary lesion to the metastatic lesion is paramount.
Elucidation of the genetic changes in the metastatic lesions compared with the matched primary tumor may help to understand the basic genetic evolutions that may be seen in the development of the metastatic lesion. The targeted genetic alterations are correlated with the driving cytogenetic abnormalities in the primary tumor and as such, investigation into the cytogenic relationships between primary and metastatic lesions may help further the targeted therapies for late metastatic lesions that can present with altered morphology, high-grade features, and challenging diagnoses.5, 6, 7, 8, 9, 10, 11 In the era of targeted therapies, this consideration has utmost importance for correctly assigning patients for enrollment in clinical trials.8, 9 Despite these implications, there is a paucity of studies examining such corresponding genetic patterns. Through the examination of the cytogenetics of metastatic deposits, we may gain an in-depth understanding of the evolution of major driver genetic mutations and drug resistance mechanisms.10, 11, 12, 13 To better understand the relationship between primary and metastatic clonal relationships in RCC, we undertook a study to examine cytogenic relationships between primary and metastatic lesions. We evaluated 39 cases of primary RCC, including clear cell, papillary, and chromophobe subtypes, and matched metastatic tumors to determine if the hallmark chromosomal anomalies of these tumors are preserved over the course of cancer progression.
Materials and methods
Thirty-nine patients with primary and metastatic RCC diagnosed between 2004 and 2014 were retrieved from the surgical pathology files. Histologic slides were reviewed, and the tumors were classified according to the 2016 World Health Organization classification system.14 The hematoxylin and eosin slides of these cases were reviewed and appropriate tumor blocks from metastatic sites were selected for cytogenetic studies. The 2010 tumor, lymph node, and metastasis (TNM) classification system was used for pathologic staging. The International Society of Urological Pathology tumor grading system was used in our study.15 For metastatic lesions, variables included location and time of presentation. Patient characteristics included gender and age. Outcome variables included subtype cytogenetic anomalies of the matched primary and metastatic tumors and their concordance. This research was approved by the Institutional Review Board.
Fluorescence in situ hybridization (FISH) analysis was performed as previously described.16, 17, 18, 19, 20 Briefly, multiple 4 μm sections were obtained from formalin-fixed paraffin-embedded tissue blocks containing neoplastic tissue. The FISH analysis was performed independently by two pathologists in a double-blinded manner without knowledge of tumor type or tumor pairing. Only one block was chosen, which was most representative of tumor. A hematoxylin and eosin-stained slide from each block was examined to identify areas of neoplastic tissue for FISH analysis. The slides were deparaffinized with two washes of xylene, 15 min each, and subsequently washed twice with absolute ethanol, 10 min each, and then air-dried in a fume hood. Next, the slides were treated with 0.1 mM citric acid (pH 6.0) (Zymed, South San Francisco, CA, USA) at 95 °C for 10 min, rinsed in distilled water for 3 min, followed by a wash of 2 × standard saline citrate (SSC) for 5 min. Tissue digestion was performed by applying 0.4 ml of pepsin (Sigma, St Louis, MO, USA) solution (4 mg/ml in 0.9% NaCl in 0.01N HCl) to each slide and incubating the slides in a humidified box for 40 min at 37 °C. The slides were rinsed with distilled water for 5 min, washed with 2 × SSC for 5 min, and then air-dried.
The alterations in chromosomes 7 and 17 were assessed using a probe cocktail containing probe CEP7 (green) and CEP17 (orange). The CEP7/CEP17 probe set and CEP Y (Green) probe were diluted with tDenHyb1 (Insitus, Albuquerque, NM, USA) in ratios of 1:50 and 1:100, respectively. Centromeric probes for chromosomes 1, 2, 6, 10, and 17 (Vysis, Downers Grove, IL, USA) were used to access the chromosome copy numbers. Each probe was diluted 1:50 in tDenHyb1 buffer (Insitus). Deletion of chromosome 3p was assessed using a probe cocktail containing BAC clone probe to chromosome 3p25 (RP11-572 M14, Green; Empire Genomics, Buffalo, NY, USA) and CEP3 (CEP3-Orange; Abbott, Downers Grove, IL, USA). The 3p25/CEP3 probe set were diluted with tDenHyb2 (Insitus) in ratios of 1:50.
The diluted probe (5 μl) was applied to each slide under reduced light conditions. The slides were then covered with a 22 × 22 mm cover slip and sealed with rubber cement. Denaturation was achieved by incubating the slides at 83 °C for 10 min in a humidified box and hybridization at 37 °C overnight. The cover slips were removed, and the slides were washed twice with 0.1 × SSC/1.5 M urea at 45 °C (20 min for each), followed by a wash with 2 × SSC for 20 min, and with 2 × SSC/0.1% NP-40 for 10 min at 45 ºC. The slides were further washed with 2 × SSC at room temperature for 5 min. The slides were air dried and counterstained with 10 μl 4,6-diamidino-2-phenylindole (Insitus), covered with cover slips, and sealed with nail polish.
The hybridized slides were observed and documented using a MetaSystem FISH system (MetaSystem, Newton, MA, USA) under × 100 oil objective. The images were acquired with a CMOS camera and analyzed with MetaSystem Isis software (MetaSystem). The following filters were used: SP-100 for DAPI, FITC MF-101 for Spectrum Green, and Gold 31003 for spectrum gold signals. Signals from each color channel (probe) were counted under false color, with computerized translation of each color channel into blue, green, and red. Four sequential focus stacks with 0.3-μm intervals were acquired and integrated into a single image to reduce thickness-related artifacts.
Analysis was performed in a manner similar to that previously described.18, 20, 21, 22, 23, 24 In brief, for each slide 100–150 nuclei from tumor tissue were scored for probe signals under the fluorescence microscope with × 1000 magnification. Definitions of chromosomal trisomy for chromosomes 7 and 17 were based on the Gaussian model and were related to the nonneoplastic renal cortex control cell signals. The cutoff values were set for each probe at the mean plus 3 standard deviations of the control values. Chromosome Y deletion was defined as ≥50% of the cells' loss of chromosome Y signal. The method of analysis for chromosomal deletions in chromophobe RCC was based on previous studies,24 and on chromosome deletions at 1p and 19q in oligodendrogliomas.25, 26 The cutoff value for chromosomal deletion was defined as ≥50% of counted cells demonstrating loss of designated signal.16, 24 The method of analysis for 3p25 deletion was based on previous studies of chromosome deletions at 1p and 19q in oligodendrogliomas.25, 26 The cutoff value for 3p deletion was defined as a 3p25/CEP3 ratio of ≤0.7. Chromosome 3p deletion was considered to be characteristic of clear cell RCC, deletion of chromosome 1, 2, 6, 10, and 17 were considered molecular features of chromophobe RCC, and trisomy of chromosomes 7 and/or 17 was considered characteristic of papillary RCC.18, 24, 27, 28
Results
The study cohort consisted of 39 patients with matched primary and metastatic RCC. The male to female ratio was 1.8:1 and the mean age was 60 years at the time of primary nephrectomy (median, range: 60, 46–82 years, respectively). Tumors were classified as clear cell RCC (n=20), papillary RCC (n=16), and chromophobe RCC (n=3), respectively. The mean primary tumor size was 9.9 cm (median, range: 10.0, 2.5–21 cm, respectively). Pathologic stage was T1 (n=2), T2 (n=6), T3 (n=30), and T4 (n=1). Sarcomatoid and rhabdoid features were present in six and two cases, respectively. At the time of nephrectomy synchronous metastases were present in 18 patients. The remainder developed metachronous metastases over a mean period of 4.1 years (median, range: 3.4, 0.6–15.8 years, respectively). The metastatic sites included bone (n=10), lymph nodes (n=10), pleura (n=1), lung (n=10), visceral (n=5), soft tissue (n=3), and mediastinum (n=1) (Table 1).
We reviewed all of the nucleolar grades for all cases and found that it showed consistent nucleolar grade between primary and metastatic tumors. Eight primary tumors had a sarcomatoid component, and the sarcomatoid component was found in four paired metastatic tumors, including two paired clear cell RCCs, one paired papillary RCC, and one paired chromophobe RCC. Although the scope of the study was not to assess the intratumoral genomic heterogeneity, it is noteworthy we did find that the cells with the chromosome 3p deletions were inconspicuous in some areas other than a few single tumor cells; it never reached a degree as to alter the chromosome 3p results.
Clear Cell RCC
A total of 20 patients with matched primary and metastatic clear cell RCC were included in the subset cohort. The male to female ratio was 1.1:1 and the mean age was 60 years at the time of primary nephrectomy (median, range: 60, 46–82 years, respectively). The mean primary tumor size was 10.2 cm (median, range: 9.8, 2.5–21 cm, respectively). Pathologic stage was T1 (n=1), T2 (n=2), and T3 (n=17). Nucleolar grade was 2 (n=6), 3 (n=9), and 4 (n=5). Sarcomatoid and rhabdoid features were present in three and two cases, respectively. Synchronous metastases were present in 8 patients and the remaining 12 patients developed metachronous metastases over a mean of 3.8 years (median, range: 3.8, 0.6–8.9 years, respectively). The metastatic sites included bone (n=7), pleura (n=1), lung (n=6), visceral (n=2), soft tissue (n=3), and mediastinum (n=1) (Table 1).
Of the 20 primary clear cell RCC tumors 18 (90%) showed a deletion of chromosome 3p. When matched for primary and metastatic site, all 20 samples displayed the same abnormality (100% concordance). All primary clear cell RCCs were disomic for chromosomes 7 and 17 (Figures 1 and 2). One patient had bilateral synchronous metastatic tumors to the lung, 0.6 and 2.0 years after primary nephrectomy. Both metastatic lesions had a 3p deletion, disomic for chromosomes 7 and 17, and were concordant with the primary tumor. Overall, there was genomic fidelity among primary and metastatic lesions regardless of clinicopathological features of the primary lesion or of the time of development or location of metastasis.

Comparison of driver chromosomal alterations between matched primary and metastatic clear cell renal cell carcinoma (RCC) and papillary RCC. The chromosomal alterations between primary and metastasis tumors showed high concordance as shown by chromosome 3p loss and trisomy 7/17 in the matched clear cell and papillary RCCs.

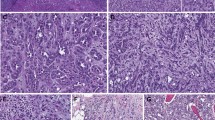
Primary and matched metastatic clear cell renal cell carcinoma. (a, b) Primary clear cell renal cell carcinoma. (b) Dual color fluorescence in situ hybridization of primary tumor demonstrating 3p deletion as indicated by the presence of a single 3p25 signal (green) with two chromosome centromere signals (red) per cell. (c, d) Matched metastatic clear cell renal cell carcinoma to the small bowel. (d) Dual color fluorescence in situ hybridization of matched metastatic tumor demonstrating 3p deletion as indicated by the presence of a single 3p25 signal (green) with two chromosome centromere signals (red) per cell.
Papillary RCC
A total of 16 patients with matched primary and metastatic papillary RCC were included in the subset cohort. The male to female ratio was 3:1 and the mean age was 61 years at the time of primary nephrectomy (median, range: 62, 46–76 years, respectively). The mean primary tumor size was 10.2 cm (median, range: 10.5, 3.5–18 cm, respectively). Pathologic stage was T1 (n=2), T2 (n=4), T3 (n=9), and T4 (n=1). Nucleolar grade was 2 (n=4), 3 (n=9), and 4 (n=3). Sarcomatoid features were present in three cases. Synchronous metastases were present in eight patients and the remaining eight patients developed metachronous metastases over a mean of 5.2 years (median, range: 4.2, 0.8–15.8 years, respectively). The metastatic sites included bone (n=2), lymph nodes (n=9), lung (n=3), and visceral (n=2) (Table 1).
In the papillary RCC cohort, 10 of the 16 papillary RCC tumors (62%) showed trisomy for both chromosomes 7 and 17 without chromosome 3p deletion. The other tumors showed disomy for both chromosomes 7 and 17 (n=4), and polysomy for both chromosomes 7 and 17 (n=2). These molecular aberrations were conserved in the 16 paired metastatic tumors (Figures 1 and 3). Y chromosome status was consistent between primary and metastatic papillary RCC. Overall, there was genomic fidelity among primary and metastatic lesions regardless of clinicopathological features of the primary lesion or of the time of development or location of metastasis.

Primary and matched metastatic papillary renal cell carcinoma. (a–c) Primary papillary renal cell carcinoma. Tumor also shows sarcomatous differentiation (a). (b) Dual color fluorescence in situ hybridization of primary tumor demonstrating trisomy of chromosomes 7 and 17 as evident by three green signals (CEP7) and three red signals (CEP7) in each nucleus. (c) Loss of Y chromosome of the primary tumor is evident by the absence of green signal in three cell nuclei. A lymphocyte retains Y signal. (d–f) Matched metastatic papillary renal cell carcinoma to the liver. (e) Dual color fluorescence in situ hybridization of matched metastatic tumor demonstrating trisomy of chromosomes 7 and 17 as evident by three green signals (CEP7) and three red signals (CEP7) in each nucleus. (f) Loss of Y chromosome of the matched metastatic tumor is evident by the absence of green signal in three cell nuclei. A lymphocyte retains Y signal.
Chromophobe RCC
A total of three patients with matched primary and metastatic chromophobe RCC were included in the subset cohort. The male to female ratio was 2:1 and the mean age was 50 years at the time of primary nephrectomy (range 47–57 years). The mean primary tumor size was 9.4 cm (range 7.4–12.2 cm, respectively). Pathologic stage was T2 (n=1) and T3 (n=2). Sarcomatoid features were present in one case. At the time of nephrectomy metastases were present in two patients. The remaining patient developed the metastatic lesion after 1.5 years. The metastatic sites included bone (n=1), lymph nodes (n=1), and visceral (n=1) (Table 1).
In the chromophobe RCC cohort, several genetic alterations were identified in chromosomes 1, 2, 6, 10, and 17 (Figure 4). Deletion of chromosome 1 was seen in one case, deletion of chromosome 6 in two cases, and deletions of chromosome 2 and 17 were seen in all cases. Overall, there was genomic fidelity among primary and metastatic lesions regardless of clinicopathological features of the primary lesion or of the time of development or location of metastasis.

Primary and matched metastatic chromophobe renal cell carcinoma. (a–c) Primary chromophobe renal cell carcinoma, low power (a) and high power (b). (c) Primary chromophobe renal cell carcinoma with losses of multiple chromosomes using interphase fluorescence in situ hybridization (FISH). FISH analysis exhibited single florescent signal for chromosome 1 (Spectrum Orange) and 6 (Spectrum Green) indicative of monosomy 1 and 6. (d–f) Matched metastatic chromophobe renal cell carcinoma to ovary, low power (d), and high power (e). (f) Matched metastatic chromophobe renal cell carcinoma with losses of multiple chromosomes using interphase FISH dual color fluorescence in situ. FISH analysis exhibited single florescent signal for chromosome 1 (Spectrum Orange) and 6 (Spectrum Green) indicative of monosomy 1 and 6.
Discussion
RCC comprises a heterogeneous group of epithelial neoplasms with diverse biological behaviors. Prognosis of RCC is limited by the development of metastases, which is present in 20–30% at the time of diagnosis. Five-year survival rates range from 50 to 85% for organ confined RCC; however, patients with distant metastases have a 5-year survival of 5–10%. The most common sites of metastatic RCC are lung, liver, and bone; however, the metastatic behavior of these epithelial neoplasms is unpredictable with often unusual sites of metastasis and with late presentation.1, 2, 3, 29
Intratumor heterogeneity may foster tumor evolution and adaptation leading to important consequences for personalized-medicine approaches that commonly rely on a single biopsy to assess the mutational landscape.30, 31, 32, 33 Recent studies have demonstrated multiple subclones found in different regions of the same tumor, suggesting evolution in a branched rather than in a linear fashion.10, 12, 34, 35, 36 In fact, the subclonal drivers represented the largest group of mutations and suggested a corresponding role in adaptation during disease development. The FISH test has the limitation that it has no global view of chromosomal alterations and only detect the chromosomal alterations that involve 1–3 MB of DNA sequence by a designated probe; it cannot detect genomic alterations involving small genetic lesions, such as point mutations and short segment gains or losses; and it is not suitable for screening cytogenetically heterogeneous diseases. However, the methodology is sensitive in detecting chromosome numerical and copy number alterations, and the types of detectable alterations tend to be stable and may evolve in a tumor's lifetime.
Additionally, recent studies have shown heterogeneity within regions of somatic mutations in primary and metastatic RCC tumors. These studies have shown significant branched evolution of the driver mutations commonly seen in clear cell RCC. However, for clear cell RCC there was also a common truncal driver alteration, which was conserved in the tested samples and showed the ubiquitous alteration in the trunk of the phylogenetic tree related to events on chromosome 3p (3p deletion or VHL mutation/methylation) during the course disease progression.34, 35, 37 Jones et al38 studied clonal divergence in clear cell RCC with sarcomatoid transformation showing a genetic divergence from a common progenitor cell. Additional studies showed that within the genetic heterogeneity of these sarcomatoid tumors, the most frequently mutated cancer drivers are those characteristically mutated in clear cell RCC. Sarcomatoid clear cell RCC harbored VHL mutations and somatic mutations linked to chromosome 3p and were shared between matched carcinomatous and sarcomatoid elements of clear cell RCC.34, 37, 39 Our study shows concordant 3p deletions between matched primary and metastatic tumors of five clear cell RCC tumors with sarcomatous features (ranging from 10 to 90% of tumor). These findings also expand the concept of genomic evolution of clear cell RCC with retained ubiquitous alterations in the trunk of the phylogenetic tree to include development of metastatic lesions from sarcomatous primary clear cell RCC. The clinical use of the cytogenetics for difficult subtyping and differential diagnosis of metastatic tumors is also supported by these findings.29
Chromosome 3p deletions are found in 70–90% of clear cell RCC, rarely found in other RCC subtypes and are considered a predominant truncal driver necessary to initiate the primary events.39 In this study we found a similar prevalence of 3p deletions in the clear cell RCC subtype cohort (90%). Similarly, 62% of in the papillary RCC subtype cohort showed trisomy of chromosome 7 and 17, which are known genetic alterations of this subtype and not commonly found in other subtypes. We did not observe aneuploidy other than trisomy 7 and 17. In the chromophobe RCC subtype cohort, genetic alterations were seen in all three tumors, which are commonly seen at chromosome 1, 2, 6, 10 13, and 17.5 The results showed 100% concordance of the driver genetic abnormalities in each tumor among all subtypes in a sizable, heterogeneous cohort of patients. The concept of conserved driver chromosomal aberrations in RCC despite the wide landscape of intratumoral genetic heterogeneity is supported by the findings of this study.34, 35, 40, 41, 42
The study supports the rationale of using cytogenetics in a metastatic RCC lesion for differential diagnosis of the primary. As aforementioned, such accurate classification is immensely important as different subtypes portend divergent prognoses and manage with disparate treatment algorithms.1, 29 The clinical use of the cytogenetics for differential diagnosis of metastatic tumors lacking primary tissue is supported by the concordance of the driver chromosomal aberrations. This can also be of use in metastatic lesions, which may present as tumors of unknown origin, or with a prior history of RCC in the absence of a known histologic subtype. In the metastatic context, fine needle aspiration specimens can be particularly challenging due to limited material for aspiration.29, 43, 44, 45 As noted in a recent study, however, FISH can often be performed using very limited tissue obtained by cytopathologic fine needle aspirations of metastatic and primary RCC tumors.46
The current study provides additional insight in the heterogenetic evolution of the metastatic lesion in clear cell RCC based on the chromosomal alterations. This study demonstrates that truncal concordance of the primary and metastatic driver genetic mutations is maintained whether metastases are synchronous or develop early or late in the course of disease (ranging from a year to decade later). These findings expand the concept of genomic evolution of RCC over a long period of metastatic tumor evolution. In other words, these metastatic tumors also maintain ubiquitous alterations in the trunk of the phylogenetic tree with loss of chromosome 3p over a long course of advanced disease evolution.34, 35, 37
This study highlights the variability in mutational signatures that can occur during the development of large renal tumors. VHL gene mutation along with 3p loss was universally found in each sample from each tumor similar to studies by Martinez et al47 demonstrating loss of 3p in 440 tumor samples from eight patients. As such, it is considered to be a truncal mutation that presents early in the phylogenetic tree found early in the tumor initiation process. The investigators found other driver mutations in the SETD2, BAP1, PBRM1, and TP53.47 However, these genes could in fact function as facultative truncal drivers if acquired early, on the other hand, such events could also occur on the 'branches' of the tree.34, 35, 37 In this regard, the genes were heterogeneously dominant driver events. Studies have also identified positive relationship between Chromosome 7 gain and MET gene copy number both type I and type II papillary RCCs in addition to MET protein expression. The loss of chromosome 3p was associated with type 2 papillary RCC and these carried frequent concurrent SETD2 mutations and mutually exclusive BAP1 and BPRM1.7, 48, 49, 50 Other data also support a parallel progression method in which individual tumors acquire diverse copy number profiles after the loss of 3p.7, 31, 33, 35 As a result, for targeted therapy of a substantial heterogeneous genomic tumor, targeting the ubiquitous aberrations may provide a logical method. If no response or partial response is found, then looking for secondary targets such as SETD2, BAP1, PBRM1, and TP53 could be employed.
The scope of our study did not include the investigation of these important genetic and chromosomal alterations, which may have a role in the molecular interplay. For example, frequencies of other (post 3p loss) genomic alterations suggest that 40–96% of these large chromosomal aberrations are heterogeneous especially if occurring late in progression. Moreover, at the current time the precise mechanisms for PBRM1, biallelic SETD2 and BAP1 tumorigenesis promotion are unknown.12 A recent study published in abstract form showed expression of BAP1 in 48 of 53 primary clear cell RCC and 18 of 19 expressions in the corresponding metastases (no clinical outcomes provided).51 Although it is a small study, the ubiquitous expression results raise questions regarding the mechanistic implications. While this area is ripe for investigation, until we have a greater understanding of the complex paracrine interactions and polygenetic resistance mechanisms of these temporal cancer subclones, targeting a dominant truncal driver may provide a more effective means of drug development.12, 31, 47
The major limitations of this study are the number of subjects. As with any retrospective study there is the potential for selection bias, recall bias, and potential impact of unknown confounders. In this study, the effects of selection and analytical bias were minimized by the blinded method of tissue selection, blinded FISH analysis, and sequential batch analysis method. The effect of recall bias and confounding variables were minimized by a single institution study with both centralized specimen procurement, and reporting and subsequent clinical follow-up.
In summary, subtyping of metastatic RCC has become increasingly important with the emergence of novel targeted therapies for specific subtypes. Additionally, as the branched tumor evolution of RCC is further explored, ubiquitous mutations on the trunk of phylogenetic tree are identified with corresponding branched tumor mutational diversity. This study demonstrated genomic fidelity among driver chromosomal alterations between matched primary and metastatic clear cell, papillary and chromophobe RCC. The findings may have important clinical and diagnostic implications for management of advanced RCC.
References
Capitanio U, Montorsi F . Renal cancer. Lancet 2015;387:894–906.
Cheng L, Williamson SR, Zhang S et al. Understanding the molecular genetics of renal cell neoplasia: implications for diagnosis, prognosis and therapy. Expert Rev Anticancer Ther 2010;10:843–864.
Frew IJ, Moch H . A clearer view of the molecular complexity of clear cell renal cell carcinoma. Annu Rev Pathol 2015;10:263–289.
Cancer Genome Atlas Research N Cancer Genome Atlas Research N Linehan WM Cancer Genome Atlas Research N Spellman PT et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 2016;374:135–145.
Shuch B, Amin A, Armstrong AJ et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol 2015;67:85–97.
Srinivasan R, Ricketts CJ, Sourbier C et al. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin Cancer Res 2015;21:10–17.
Linehan WM, Ricketts CJ . Decade in review-kidney cancer: discoveries, therapies and opportunities. Nat Rev Urol 2014;11:614–616.
Powles T, Staehler M, Ljungberg B et al. Updated EAU guidelines for clear cell renal cancer patients who fail VEGF targeted therapy. Eur Urol 2016;69:4–6.
Albiges L, Choueiri T, Escudier B et al. A systematic review of sequencing and combinations of systemic therapy in metastatic renal cancer. Eur Urol 2015;67:100–110.
Brugarolas J . Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol 2014;32:1968–1976.
Incorvaia L, Bronte G, Bazan V et al. Beyond evidence-based data: scientific rationale and tumor behavior to drive sequential and personalized therapeutic strategies for the treatment of metastatic renal cell carcinoma. Oncotarget [serial online] 2016. Available: http://www.ncbi.nlm.nih.gov/pubmed/26872372 [accessed 8 February 2016).
Ricketts CJ, Linehan WM . Intratumoral heterogeneity in kidney cancer. Nat Genet 2014;46:214–215.
Chen F, Zhang Y, Senbabaoglu Y et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep 2016;14:2476–2489.
Moch HHP, Ulbright TM, Reuter VE . WHO Classification of Tumours of the Urinary System and Male Genital Organ. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2016.
Delahunt B, Egevad L, Montironi R et al. International Society of Urological Pathology (ISUP) consensus conference on renal neoplasia: rationale and organization. Am J Surg Pathol 2013;37:1463–1468.
Brunelli M, Gobbo S, Cossu-Rocca P et al. Fluorescent cytogenetics of renal cell neoplasms. Pathologica 2008;100:454–460.
Cossu-Rocca P, Eble JN, Delahunt B et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006;19:488–493.
Gobbo S, Eble JN, Grignon DJ et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008;32:1239–1245.
Halat S, Eble JN, Grignon DJ et al. Multilocular cystic renal cell carcinoma is a subtype of clear cell renal cell carcinoma. Mod Pathol 2010;23:931–936.
Williamson SR, Gupta NS, Eble JN et al. Clear cell renal cell carcinoma with borderline features of clear cell papillary renal cell carcinoma: combined morphologic, immunohistochemical, and cytogenetic analysis. Am J Surg Pathol 2015;39:1502–1510.
Cheng L, MacLennan GT, Zhang S et al. Evidence for polyclonal origin of multifocal clear cell renal cell carcinoma. Clin Cancer Res 2008;14:8087–8093.
Gobbo S, Eble JN, Maclennan GT et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 2008;32:1780–1786.
Williamson SR, Zhang S, Eble JN et al. Clear cell papillary renal cell carcinoma-like tumors in patients with von Hippel-Lindau disease are unrelated to sporadic clear cell papillary renal cell carcinoma. Am J Surg Pathol 2013;37:1131–1139.
Brunelli M, Eble JN, Zhang S et al. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol 2005;18:161–169.
Weller M, Berger H, Hartmann C et al. Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin Cancer Res 2007;13:6933–6937.
Wiens AL, Cheng L, Bertsch EC et al. Polysomy of chromosomes 1 and/or 19 is common and associated with less favorable clinical outcome in oligodendrogliomas: fluorescent in situ hybridization analysis of 84 consecutive cases. J Neuropathol Exp Neurol 2012;71:618–624.
Cheng L, Zhang S, MacLennan GT et al. Molecular and cytogenetic insights into the pathogenesis, classification, differential diagnosis, and prognosis of renal epithelial neoplasms. Hum Pathol 2009;40:10–29.
Brunelli M, Eble JN, Zhang S et al. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol 2003;16:1053–1059.
Wang L, Williamson SR, Wang M et al. Molecular subtyping of metastatic renal cell carcinoma: implications for targeted therapy. Mol Cancer 2014;13:39.
Pon JR, Marra MA . Driver and passenger mutations in cancer. Annu Rev Pathol 2015;10:25–50.
McGranahan N, Swanton C . Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015;27:15–26.
Catenacci DV . Next-generation clinical trials: novel strategies to address the challenge of tumor molecular heterogeneity. Mol Oncol 2015;9:967–996.
Alizadeh AA, Aranda V, Bardelli A et al. Toward understanding and exploiting tumor heterogeneity. Nat Med 2015;21:846–853.
Gerlinger M, Rowan AJ, Horswell S et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–892.
Gerlinger M, Horswell S, Larkin J et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014;46:225–233.
Durinck S, Stawiski EW, Pavia-Jimenez A et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 2015;47:13–21.
Bi M, Zhao S, Said JW et al. Genomic characterization of sarcomatoid transformation in clear cell renal cell carcinoma. Proc Natl Acad Sci USA 2016;113:2170–2175.
Jones TD, Eble JN, Wang M et al. Clonal divergence and genetic heterogeneity in clear cell renal cell carcinomas with sarcomatoid transformation. Cancer 2005;104:1195–1203.
Malouf GG, Ali SM, Wang K et al. Genomic characterization of renal cell carcinoma with sarcomatoid dedifferentiation pinpoints recurrent genomic alterations. Eur Urol 2016;70:348–357.
Gronwald J, Storkel S, Holtgreve-Grez H et al. Comparison of DNA gains and losses in primary renal clear cell carcinomas and metastatic sites: importance of 1q and 3p copy number changes in metastatic events. Cancer Res 1997;57:481–487.
Bissig H, Richter J, Desper R et al. Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am J Pathol 1999;155:267–274.
Sharpe K, Stewart GD, Mackay A et al. The effect of VEGF-targeted therapy on biomarker expression in sequential tissue from patients with metastatic clear cell renal cancer. Clin Cancer Res 2013;19:6924–6934.
Volpe A, Finelli A, Gill IS et al. Rationale for percutaneous biopsy and histologic characterisation of renal tumours. Eur Urol 2012;62:491–504.
Wilkerson ML, Lin F, Liu H et al. The application of immunohistochemical biomarkers in urologic surgical pathology. Arch Pathol Lab Med 2014;138:1643–1665.
Patel HD, Johnson MH, Pierorazio PM et al. Diagnostic accuracy and risks of biopsy in the diagnosis of a renal mass suspicious for localized renal cell carcinoma: systematic review of the literature. J Urol 2016;195:1340–1347.
Kos Z, Williams PA, Belanger EC et al. Fluorescence in situ hybridization as an adjunct tool in the diagnosis of primary and metastatic renal cell carcinoma in fine needle aspiration specimens. Diagn Cytopathol 2014;42:1013–1023.
Martinez P, Birkbak NJ, Gerlinger M et al. Parallel evolution of tumour subclones mimics diversity between tumours. J Pathol 2013;230:356–364.
Yin X, Zhang T, Su X et al. Relationships between chromosome 7 gain, MET gene copy number increase and MET protein overexpression in Chinese papillary renal cell carcinoma patients. PLoS One 2015;10:e0143468.
Yu M, Liang H, Fu Z et al. BAP1 suppresses lung cancer progression and is inhibited by miR-31. Oncotarget 2016;7:13742–13753.
Pfister SX, Ahrabi S, Zalmas LP et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep 2014;7:2006–2018.
Freiedman P . Immunohistochemical comparison of BAP1 immunoexpression status between needle biopsies and nephrectomies in clear cell renal cell carcinoma. Mod Pathol 2016;25:229a.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kouba, E., Eble, J., Simper, N. et al. High fidelity of driver chromosomal alterations among primary and metastatic renal cell carcinomas: implications for tumor clonal evolution and treatment. Mod Pathol 29, 1347–1357 (2016). https://doi.org/10.1038/modpathol.2016.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.133
