
Abstract
The effects of nephropathy risk variants in the apolipoprotein L1 gene (APOL1) on renal histopathology in African Americans with arterionephrosclerosis or putative ‘hypertension-associated’ nephropathy are unknown. APOL1 genotype–phenotype correlations were performed in a blinded manner from renal biopsies in 196 self-reported African Americans with arterionephrosclerosis on kidney biopsy at a large national nephropathology practice. Subjects had chronic kidney disease without nephrotic syndrome. A discovery analysis compared histopathologic changes in the glomerular and tubulointerstitial compartments in 58 subjects with 2 versus 56 subjects with 0 APOL1 risk variants. Validation was performed in biopsies from 82 additional subjects with 0, 1, and 2 risk variants. Two risk variant versus zero risk variant group genotype associations and subphenotypes were assessed by χ2 analyses. ANOVA compared means of continuous variables. In discovery analyses, significantly less obsolescent glomerulosclerosis, more (solidified and disappearing) glomerulosclerosis, more thyroidization-type tubular atrophy, and more microcystic tubular dilatation were seen in patients with two versus zero APOL1 risk alleles. Greater degrees of arteriosclerosis were present in those with zero risk alleles. Segmental glomerulosclerosis did not differ significantly between groups. Presence of two of the following discriminatory histopathologic findings from discovery, that is, <50% obsolescent glomerulosclerosis, thyroidization-type tubular atrophy, and microcystic tubular dilatation, was specific for the presence of two APOL1 risk alleles in the validation phase. African Americans with arterionephrosclerosis who possess two APOL1 risk variants more often lack obsolescent glomerulosclerosis and have greater degrees of (solidified and disappearing) glomerulosclerosis, thyroidization-type tubular atrophy, and microcystic tubular dilation than patients with fewer than two risk variants. These findings support involvement of multiple cell types in subnephrotic forms of APOL1-associated nephropathy, particularly renal tubule cells with resultant tubulointerstitial disease.
Similar content being viewed by others


Main
Compared with populations who lack recent African ancestry, African Americans have higher incidence rates of chronic kidney disease (CKD).1 Two coding risk variants in the apolipoprotein L1 gene (APOL1), termed G1 and G2, are responsible for much of the increased incidence of progressive, nondiabetic kidney disease in African Americans relative to European Americans (EAs).2, 3 The spectrum of APOL1-associated kidney disease includes focal segmental glomerulosclerosis, nondiabetic end-stage kidney disease often attributed to systemic hypertension, collapsing glomerulopathy in the settings of systemic lupus erythematosus4, 5 and HIV,6 and sickle cell nephropathy.7 The pathogenic mechanisms by which these risk variants produce disease are unknown. Discovery is complicated by the lack of animal models of disease, as the APOL1 gene is limited to humans and a few higher level primates.8 Therefore, careful renal histologic observations in patients with clinical nephropathy are imperative.
CKD has traditionally been attributed to ‘hypertensive nephrosclerosis’ when the clinical history includes hypertension and slowly progressive renal failure without other comorbid illnesses.9, 10 The African American Study of Kidney Disease and Hypertension (AASK) demonstrated that APOL1 risk variants were strongly associated with hypertension-attributed CKD in a recessive model with an odds ratio of 2.57; however, only 23% of AASK participants possessed 2 APOL1 risk variants; thus, cases with non-APOL1-mediated arteriolar nephrosclerosis were included in the study.11 It is unknown whether APOL1-associated CKD has unique histopathology compared with ordinary (non-APOL1-mediated) forms of hypertensive nephrosclerosis.12 Only one study has compared the morphologic findings in African Americans and EAs with putative hypertensive nephrosclerosis; this study showed an increase in the solidified pattern of glomerulosclerosis in African-American kidney biopsies.13 However, the study failed to account for APOL1 risk alleles in the African-American population as it was performed before discovery of APOL1-associated nephropathy in 2010. Therefore, we performed an APOL1 genotype–phenotype correlation in kidney biopsies from African Americans who had hypertension-attributed CKD without the nephrotic syndrome.
Materials and methods
Participant Inclusion Criteria
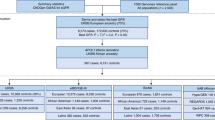
All kidney biopsies in self-reported African Americans who had a clinical diagnosis of CKD stage ≥3 (estimated glomerular filtration rate (eGFR) <60 ml/min per 1.73 m2 for at least 3 months) with a diagnosis of arterionephrosclerosis made at Nephropath between 2010 and 2012 were identified. The diagnosis of arterionephrosclerosis was based on clinicopathologic findings including a history of hypertension and progressive renal failure without other comorbid illnesses as well as nonspecific chronic injury changes on kidney biopsy including arteriosclerosis. Figure 1 includes a flow diagram displaying case selection for discovery and validation analyses. Genotyping of single-nucleotide polymorphisms (SNPs) in APOL1 was performed if at least eight glomeruli were present for light microscopic evaluation. Patients were excluded if they had nephrotic syndrome (24 h urine protein >3.5 g per day, hypoalbuminemia (<3.5 g/dl), and peripheral edema) as it was suggestive of primary focal segmental glomerulosclerosis and podocytopathy, a history of diabetes mellitus, or any systemic diseases associated with CKD except for systemic hypertension. Cases with proteinuria ≥3.5 g per day were not excluded in the absence of the nephrotic syndrome. This study was approved by the Institutional Review Boards of the University of Arkansas for Medical Sciences.

Flow diagram for case selection during the discovery and validation phases.
Genotyping
DNA from frozen kidney tissue was genotyped for APOL1 risk alleles using TaqMan assays.4 The G1S342G (rs73885319) and G2 insertion/deletion (rs71785313) were typed to assess kidney disease risk. Genome-wide ancestry informative markers were not genotyped as they have not significantly altered association results beyond G1 and G2 in prior reports.6
Study Design and Histologic Grading of Renal Biopsies
The discovery analysis was performed in biopsies from individuals who had either two or zero APOL1 risk alleles. This analysis was designed to provide maximum discrimination between the effects of APOL1 risk variants on renal histology. A subsequent validation analysis was performed by four nephropathologists on biopsies from individuals with two, one, or no APOL1 risk variants. This analysis included individuals with a single risk variant to assess whether their histology differed from those with zero or two risk variants. Pathologists were blinded to the APOL1 genotype during morphologic evaluation.
Globally sclerotic glomeruli were characterized as obsolescent, solidified, or disappearing, as previously reported.13 Sclerotic glomeruli were considered obsolescent when the sclerotic glomerular tuft was retracted with accumulation of fibrous matrix replacing Bowman’s space (Figure 2). The solidified pattern of glomerulosclerosis reflected a solidified sclerotic tuft without retraction and without a fibrous matrix surrounding the tuft. The disappearing pattern of glomerulosclerosis is characterized by partial or complete disappearance of Bowman’s capsule with the sclerotic glomerular tuft beginning to blend with the background fibrosis. The disappearing form of glomerulosclerosis frequently has inflammatory cells infiltrating the sclerotic tuft. Cases with segmental glomerulosclerosis were classified according to the Columbia classification.14

Renal biopsy findings in African Americans with hypertension, nephrosclerosis, and stage ≥3 CKD. (a) Tubules with classic-type tubular atrophy show thickened and wrinkled basement membranes with occasional basement membrane lamellation (periodic acid–Schiff, original magnification × 400). (b) Tubules with thyroidization-type atrophy have a luminal hyaline cast surrounded by simplified epithelium (periodic acid–Schiff, original magnification × 200). (c) Endocrine-type tubular atrophy with narrow or no tubular lumen, clear cytoplasm, and thin basement membranes (periodic acid–Schiff, original magnification × 400). (d) Glomerulus showing ischemic obsolescence with contracted sclerotic tuft and surrounding fibrosis in Bowman’s space surrounded by intact Bowman’s capsule (periodic acid–Schiff, original magnification × 400). (e) Disappearing pattern of glomerulosclerosis with complete disappearance of Bowman’s capsule and the sclerotic glomerular tuft beginning to blend with background fibrosis (periodic acid–Schiff, original magnification × 400). (f) Solidified glomerulosclerosis in which the sclerotic tuft is not contracted and fills Bowman’s space (periodic acid–Schiff, original magnification × 400).
Tubular atrophy was scored as: none (0) when <10% of the renal cortex tubules were atrophic, mild (1) when 10–25% of the cortical tubules were atrophic, moderate (2) when 26–50% of the cortical tubules were atrophic, and severe (3) when >50% of the cortical tubules were atrophic. Tubular atrophy was further characterized into three types including classic-type, endocrine-type, and thyroidization-type tubular atrophy (Figure 2). The degree of each type of atrophy was scored using the same criteria used for total atrophy. Therefore, the total score of the tubular atrophy subtypes always equaled the total tubular atrophy score. Classic tubular atrophy was used when the atrophic tubules had thickened, wrinkled, and sometimes lamellated basement membranes with a simplified epithelium. Thyroidization-type tubular atrophy was scored when the tubular epithelium was flattened and the tubular lumen contained a rounded hyaline protein cast. Endocrine-type tubular atrophy was scored when the atrophic tubule was small with narrow or absent lumen and clear regular epithelial cells.15 Microcystic tubular dilatation was defined as dilatation of a tubular lumen to at least three times the diameter of a normal tubular profile with a large intraluminal PAS-positive hyaline cast.
Definitions for degree of interstitial fibrosis, tubular atrophy, interstitial inflammation, tubulitis, mesangial expansion, endocapillary hypercellularity, arteriolar hyalinosis, arterial intimal fibrosis, and glomerular basement membrane duplication were scored according to the definitions provided in the Banff working classification.16 Degree of glomerular basement membrane thickening was scored as: 0, within 1 s.d. of mean; 1, >1 and <2 s.d. of mean; 2, >2 and <3 s.d. of mean; 3, >3 s.d. above mean. Degree of foot process effacement was scored based on a visual estimate as follows: 0, effacement involving <10% of glomerular basement membrane surface area; 1, effacement involving 10–30% of glomerular basement membrane surface area; 2, effacement involving 30–60% of glomerular basement membrane surface area; 3, effacement involving >60% of glomerular basement membrane surface area. The presence of glomerulomegaly was determined based on light microscopic inspection of glomeruli in each sample cut at or near the hilum.
Statistical Analyses
Cases with two APOL1 risk variants versus controls with zero APOL1 risk variants had subphenotypes assessed by χ2 analyses. ANOVA was used to compare means of continuous variables. The Mann–Whitney test was used to compare ordinal data. We considered nominal P-values of <0.05 as significant for all tests.
Results
Regardless of genotype, kidney biopsies in all subjects revealed nonspecific chronic tubulointerstitial injury and arteriosclerosis that historically would have been considered consistent with arterionephrosclerosis. The morphologic analyses were divided into two stages. A discovery phase included 114 biopsies including 58 biopsies with 2 risk alleles and 56 biopsies with 0 risk alleles. Electron microscopy was evaluated in this phase comparing 42 patients in the 0 risk allele group with those seen in 44 patients with 2 risk alleles. The validation phase consisted of 82 cases including 23 with 0 risk alleles, 25 with 1 risk allele, and 34 with 2 risk alleles.
The results of the morphologic evaluation carried out in the discovery phase are listed in Table 1. Focal segmental glomerulosclerosis lesions were very common with no significant difference between the two genotype groups. There were 44 (79%) cases with 0 risk alleles who had focal segmental glomerulosclerosis lesions compared with 43 (74%) with 2 risk alleles. The most common variant of focal segmental glomerulosclerosis was not otherwise specified (39/44 with 0 risk alleles and 41/43 with 2 risk alleles), followed by the perihilar variant (5/44 with 0 risk alleles and 2/43 with 2 risk alleles). There were no cases of cellular, collapsing, or tip variant. There were no cases in either genotype group with endocapillary proliferation or crescent formation. There was no significant difference in the degree of glomerular basement membrane thickening by electron microscopy in the two groups and no cases with glomerular basement membrane thinning were present in either group.
Following the discovery phase, the morphologic findings that were most significantly associated with the presence of APOL1 risk alleles were evaluated in a validation set of consecutive biopsies from African-American patients who met the inclusion criteria. There were 82 cases in this validation phase including 23 with 0 risk alleles, 25 with 1 risk allele, and 34 with 2 risk alleles. Results of this analysis are listed in Table 2.
The three morphologic features most strongly associated with two APOL1 risk alleles in discovery were evaluated in validation samples to test the sensitivity and specificity of the findings for APOL1-associated kidney disease in African Americans with CKD. Morphologic findings selected for inclusion in the scoring system were <50% globally sclerotic glomeruli of the obsolescent type, >1 focus of microcystic tubular dilatation, and >50% of total tubular atrophy of thyroidization type. Applying these three criteria to the discovery phase, the presence of at least two criteria had a sensitivity of 43% and a specificity of 92% for presence of two APOL1 risk variants. These morphologic features were scored by four renal pathologists with experience ranging from beginner (<1 year of experience) to highly experienced (>30 years of experience) in the validation phase. In this phase, the presence of two of these three had a mean sensitivity among the four pathologists of 36% and specificity of 86% for presence of two APOL1 risk variants. Interobserver agreement among the renal pathologists was good for identifying cases with two risk variants (at least two of these histologic criteria) with a κ of 0.71.
The discovery and validation cohorts were combined for a final analysis with all cases (Table 3). Serum creatinine concentrations and degree of proteinuria (where available) were included in this analysis and reflect fairly advanced kidney disease. The results of the combined analysis are similar to the two independent analyses. No significant differences were seen in the serum creatinine or degree of proteinuria between the risk allele groups.
Discussion
As in the AASK, morphologic findings in kidney biopsies from hypertensive, nondiabetic African Americans with CKD who lack nephrotic syndrome are generally nonspecific.17 These injury patterns represent final common pathways for multiple forms of renal disease. Kidney biopsies with these findings are commonly diagnosed as ‘hypertensive’ or arteriolar nephrosclerosis in the absence of systemic diseases other than hypertension that associate with nephropathy. Many such cases of hypertension-attributed kidney disease in African Americans are now known to be manifestations of APOL1-associated nephropathy.11, 18 The present analyses are the first to assess whether there are histopathologic findings in these renal biopsies that more frequently associate with APOL1 risk alleles. Solidified and disappearing glomerulosclerosis, mean degrees of interstitial fibrosis, tubular atrophy (specifically thyroidization-type tubular atrophy), and percentage of cases with microcystic tubular dilatation were all significantly greater in subjects with two APOL1 risk variants, relative to those with zero. In contrast, obsolescent glomerulosclerosis and greater degrees of arteriosclerosis were present significantly more often in those without risk APOL1 variants. These results extend observations by Marcantoni et al13 regarding the different renal histology in African Americans and EAs diagnosed with arteriosclerosis. In retrospect, population ancestry-based differences in many African-American subjects from that report are likely related to APOL1 risk variants; variants that are virtually absent in EAs. In fact, our report suggests that the renal histology in African Americans lacking two APOL1 risk variants is quite similar to EAs from the report of Marcantoni et al.13
Microcystic tubular dilatation was pronounced in subjects with two APOL1 risk variants in the present report. It is noteworthy that microcystic tubular dilation is increased in almost all forms of APOL1-associated kidney disease, including HIV-associated nephropathy,19 lupus-associated collapsing glomerulopathy,4 and now CKD with arterionephrosclerosis. Even when dilatation of the tubular lumens was absent, there was often a marked increase in the presence of tubular casts in APOL1 two risk variant disease in the form of thyroidization-type tubular atrophy. Though once thought to be specific for chronic pyelonephritis,20 hyaline casts are now known to be common in areas of chronicity of all types. They are thought to be an amalgam of various filtered proteins with Tamm–Horsfall protein.21 However, detailed studies have not been performed to determine the exact composition of these casts. As such, it is not known if the compositions of casts in patients with APOL1 risk alleles are unique. These morphologic findings support a major role for renal tubule cell injury in the pathogenesis of several forms of APOL1-associated nephropathy.
APOL1 message and protein are expressed in arteriolar endothelial cells in normal kidneys.22 However, the staining distribution may vary in patients with focal segmental glomerulosclerosis or HIV-associated nephropathy such that there is marked increased in APOL1 staining in the vessel walls rather than the vascular endothelium.22 These findings have raised speculation that APOL1 risk variants might have a pathogenic role in promoting arteriosclerosis.12 In addition, the fact that medium-sized HDL subclass concentrations decrease for each additional APOL1 risk variant may support a vasculocentric pathogenesis for APOL1-mediated kidney disease.23 Decreases in medium-sized HDL particle concentrations have been shown to contribute to coronary artery disease and could have a similar effect on the renal microvasculature.24 If APOL1 risk variants lead to microvascular alterations that in turn lead to hypertension and renal failure, one would expect to see severe arteriosclerosis in these cases. However, we show that the majority of the APOL1 cases did not have severe vascular changes. In fact, there was a trend toward less arteriosclerosis in cases with two risk alleles. In addition, glomerular obsolescence, the pattern of glomerular sclerosis resulting from reduced glomerular perfusion caused by arteriosclerosis of the small arteries in the kidney,25 is significantly decreased in the two risk allele group compared with those with no risk alleles. Given these findings, vascular abnormalities may not be the pathogenic drivers of APOL1-related CKD in those with low-level proteinuria.
The most aggressive form of APOL1-related nephropathy is collapsing glomerulopathy. This form of glomerulopathy has been classified as a podocytopathy in which injury to the podocyte is the primary mechanism of renal disease. Heavy proteinuria is often present. Similarly, primary focal segmental glomerulosclerosis, which also resides in the APOL1 disease spectrum, is considered by many to be driven by podocyte injury. We sought to remove cases of primary focal segmental glomerulosclerosis from this study by eliminating cases with nephrotic syndrome. Although podocytopathy might play a role in the collapsing and focal segmental glomerulosclerosis varieties of APOL1 disease, the findings in this study do not support the concept that ongoing podocyte injury plays a significant role in hypertension-attributed CKD associated with APOL1. Epithelial foot process effacement, considered to be morphologic evidence of podocyte injury, was less prominent in the patients with two risk alleles than in the patients with no risk alleles. In addition, there was no significant difference between the degrees of focal segmental glomerulosclerosis in cases with two, one, or zero risk alleles. However, it should be pointed out that the degree of segmental sclerosis was significantly higher in the zero risk allele group in the discovery phase relative to the validation phase. The reasons for this discrepancy are uncertain. One possible explanation is that less chronicity was present in the no risk allele group in the validation phase compared with that in the discovery phase. These morphologic findings suggest that podocytes may not be the primary cells involved in APOL1-associated CKD with low-level proteinuria; however, this cannot be concluded solely based on morphology.
Finally, we sought to provide a means by which renal pathologists could differentiate APOL1-associated CKD from arterionephrosclerosis in hypertensive, nondiabetic African-American subjects. We have shown that the presence of two out of three possible histopathologic findings accomplishes the goal of being highly specific for identifying patients with APOL1 risk alleles; it is also very reproducible from beginner to highly experienced pathologists. The sensitivity for identifying patients with APOL1 risk alleles among patients with CKD is quite low. However, this is not surprising and does not necessarily indicate low sensitivity for APOL1 disease. It is well known that most patients with APOL1 risk alleles do not develop nephropathy.26 As there is a low risk for CKD in those with two APOL1 risk alleles (potentially requiring a second hit for progression of disease), it is likely that many patients with two risk alleles but without a second hit have renal disease that is pathogenically similar to patients without risk alleles. In these cases, we would expect the morphologic findings to correspond more closely to the no risk allele group despite presence of risk alleles. However, we cannot exclude the possibility that many more patients have APOL1-associated CKD than we are detecting with this system because of the insensitivity of the morphologic findings. The scoring system was designed and validated to evaluate African-American patients with CKD and biopsy findings otherwise considered diagnostic of arterionephrosclerosis. The specificity seen in this setting might not be present in other clinicopathologic scenarios.
This study has several strengths and also limitations. A relatively large number of biopsies were analyzed. However, detailed clinical data were not always available as the samples were collected by a pathology practice. Variable information is often provided by referring clinicians. In addition, genotyping for ancestry informative markers was not performed as adjustment for these markers have not markedly affected results in other reports of APOL1-associated kidney disease. The patients with two APOL1 risk alleles tended to be younger than those with less than two risk alleles. Consequently, we cannot exclude the possibility that some of the changes identified are age related and APOL1 is serving as a nephropathy progression factor. Finally, relatively few subjects with one risk variant were analyzed as the discovery phase compared two risk variants with none to maximize the chance to identify unique histologic alterations in those with two variants. The validation phase included subjects with one risk variant. APOL1-associated nephropathy is inherited in an autosomal recessive manner and no major histologic differences were seen between those with zero or one risk variant.
We present the first detailed morphologic investigation of the histopathology of APOL1-associated CKD. This study demonstrates higher frequencies of solidified and disappearing glomerulosclerosis, higher mean degrees of interstitial fibrosis, tubular atrophy specifically of the thyroidization type, and higher percentage of cases with microcystic tubular dilatation in African Americans with two APOL1 risk variants. In those without risk APOL1 variants, obsolescent glomerulosclerosis and greater degrees of arteriosclerosis were more often present. The histopathologic changes in APOL1-associated nephropathy were present in every compartment of the kidney. A simple scoring system may make it possible to identify cases suspicious for APOL1 risk alleles based on kidney biopsy findings. Although many of these morphologic changes are nonspecific, when they are present together they have a high specificity for APOL1-associated kidney disease in African Americans with hypertension and CKD. These biopsy findings may prompt clinicians to genotype African-American patients for presence of two APOL1 G1 and G2 risk variants. This would signify high risk for progression of nephropathy and may prove to be important in the selection of living related kidney donors from their families.18
References
Easterling RE . Racial factors in the incidence and causation of end-stage renal disease (ESRD). Trans Am Soc Artif Intern Organs 1977;23:28–33.
Genovese G, Friedman DJ, Ross MD et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010;329:841–845.
Tzur S, Rosset S, Shemer R et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010;128:345–350.
Larsen CP, Beggs ML, Saeed M et al. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol 2013;24:722–725.
Freedman BI, Langefeld CD, Andringa KK et al. End-stage kidney disease in African Americans with lupus nephritis associates with APOL1. Arthritis Rheumatol 2014;66:390–396.
Kopp JB, Nelson GW, Sampath K et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 2011;22:2129–2137.
Ashley-Koch AE, Okocha EC, Garrett ME et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol 2011;155:386–394.
Friedman DJ, Pollak MR . Genetics of kidney failure and the evolving story of APOL1. J Clin Invest 2011;121:3367–3374.
Skorecki KL, Wasser WG . Hypertension-misattributed kidney disease in African Americans. Kidney Int 2012;83:9–13.
Freedman BI, Sedor JR . Hypertension-associated kidney diseases: perhaps no more. J Am Soc Nephrol 2008;19:2047–2051.
Lipkowitz MS, Freedman BI, Langefeld CD et al. Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int 2012;83:114–120.
Kopp JB . Rethinking hypertensive kidney disease:arterionephrosclerosis as a genetic, metabolic, and inflammatory disorder. Curr Opin Nephrol Hypertens 2013;22:266–272.
Marcantoni C, Ma LJ, Federspiel C et al. Hypertensive nephrosclerosis in African Americans versus Caucasians. Kidney Int 2002;62:172–180.
D'Agati VD, Fogo A, Bruijn JA et al. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 2004;43:368–382.
Nadasdy T, Laszik Z, Blick KE et al. Tubular atrophy in the end-stage kidney: a lectin and immunohistochemical study. Hum Pathol 1994;25:22–28.
Racusen LC, Solez K, Colvin RB et al. The Banff 97 working classification of renal allograft pathology. Kidney Int 1999;55:713–723.
Fogo A, Breyer JA, Smith MC et al. Accuracy of the diagnosis of hypertensive nephrosclerosis in african americans: a report from the African American Study of Kidney Disease (AASK) trial. Kidney Int 1997;51:244–252.
Parsa A, Kao WH, Xie D et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013;1–14.
Laurinavicius A, Hurwitz S, Rennke HG . Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int 1999;56:2203–2213.
Laberke HG, Klingebiel T, Quack G . A contribution to the morphology and pathogenesis of thyroid-like lesions in the kidney. Path Res Pract 1983;176:284–296.
Cohen AH . Morphology of renal tubular hyaline casts. Lab Invest 1981;44:280–287.
Madhavan SM, O’Toole JF, Konieczkowski M et al. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 2011;22:2119–2128.
Freedman BI, Langefeld CD, Murea M et al. Apolipoprotein L1 nephropathy risk variants associate with HDL subfraction concentration in African Americans. Nephrol Dial Transplant 2011;26:3805–3810.
Kuller LH, Grandits G, Cohen JD et al. Lipoprotein particles, insulin, adiponectin, C-reactive protein and risk of coronary heart disease among men with metabolic syndrome. Atherosclerosis 2007;195:122–128.
Hughson MD, Johnson K, Young RJ et al. Glomerular size and glomerulosclerosis: relationships to disease categories, glomerular solidification, and ischemic obsolescence. Am J Kidney Dis 2002;39:679–688.
Foster MC, Coresh J, Fornage M et al. APOL1 variants associate with increased risk of CKD among african americans. J Am Soc Nephrol 2013;24:1484–1491.
Acknowledgements
Dr Freedman is supported by NIH RO1 DK070941 and DK084149.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Larsen, C., Beggs, M., Saeed, M. et al. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod Pathol 28, 95–102 (2015). https://doi.org/10.1038/modpathol.2014.92
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.92
This article is cited by
-
COVID-19 associated collapsing glomerulopathy presenting as acute kidney injury on chronic kidney disease: a case report and review of the literature
CEN Case Reports (2022)
-
APOL1 genotype-associated morphologic changes among patients with focal segmental glomerulosclerosis
Pediatric Nephrology (2021)
-
Lessons for the clinical nephrologist: recurrence of nephrotic syndrome induced by SARS-CoV-2
Journal of Nephrology (2020)
-
A mouse recapitulating APOL1-associated kidney disease
Nature Medicine (2017)
-
Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice
Nature Medicine (2017)
