
Abstract
Heredity is a major cause of colorectal cancer, but although several rare high-risk syndromes have been linked to disease-predisposing mutations, the genetic mechanisms are undetermined in the majority of families suspected of hereditary cancer. We review the clinical presentation, histopathologic features, and the genetic and epigenetic profiles of the familial colorectal cancer type X (FCCTX) syndrome with the aim to delineate tumor characteristics that may contribute to refined diagnostics and optimized tumor prevention.
Similar content being viewed by others


Main
Heredity represents a major cause of colorectal cancer, with at least 20% of the cases estimated to develop because of genetic factors and ∼5% linked to inherited mutations in cancer-predisposing genes.1, 2, 3, 4 Worldwide, hereditary factors are annually estimated to cause 100 000 deaths from colorectal cancer. Patients whose tumors are identified at early stages have an excellent prognosis, and this implies an unprecedented possibility for disease prevention in individuals at increased risk. Regular colonoscopies have been demonstrated to effectively reduce morbidity and mortality from colorectal cancer in individuals with hereditary predisposition for the disease.5, 6
Hereditary colorectal cancer can broadly be divided into polyposis syndromes and nonpolyposis syndromes. The polyposis subset represents <1% of the cases and includes, for example, familial adenomatous polyposis (FAP) caused by ‘APC (adenomatous polyposis coli)’ mutations, MUTYH-associated polyposis (MAP) caused by ‘MUTYH (mutY homolog)’ mutations, Peutz–Jegher syndrome with ‘STK11 (serine/threonine kinase 11)’ mutations, and Juvenile Polyposis with mutations in ‘SMAD4 (SMAD family member 4)’ and ‘BMPR1A (bone morphogenetic protein receptor, type IA).’7, 8, 9, 10, 11, 12, 13 The term hereditary nonpolyposis colorectal cancer (HNPCC) was coined to distinguish familial aggregation of colorectal cancer from the polyposis phenotypes. The Amsterdam criteria (AC) were introduced for uniform classification based on family history and require at least three affected family members in two or more generations, with one being a first-degree relative of the other two and at least one individual diagnosed before 50 years of age.14, 15 The AC1 apply to families with ≥3 colorectal cancers and the AC2 also include extracolonic tumors, that is, endometrial cancer, cancer of the upper urinary tract, and cancer of the small bowel.14, 15 The AC are fulfilled in 2–5% of patients with colorectal cancer.4, 16, 17 The HNPCC subset of colorectal cancer families is heterogeneous and broadly consists of the 4% linked to Lynch syndrome (which may or may not fulfill the AC), <1% with a Lynch-like syndrome, and 2–4% classified as familial colorectal cancer type X (FCCTX). Lynch syndrome is defined by germline mismatch-repair (MMR) gene mutations in ‘MLH1 (mutL homolog 1),’ ‘MSH2 (mutS homolog 2),’ ‘MSH6 (mutS homolog 6), ‘PMS2 (PMS2 postmeiotic segregation increased 2, S. cerevisiae)’, but only about one-third of the Lynch syndrome families fulfill the AC criteria.18, 19 Lynch-like families are defined by the AC and show tumors with functional MMR gene defects (ie, loss of MMR protein expression and/or presence of microsatellite instability (MSI)), but lack of disease-predisposing MMR gene mutations.20 FCCTX families are defined as families that fulfill the AC1 and show MMR-stable tumors and lack of MMR gene mutations.4, 16, 19, 21 Some studies of FCCTX have also included AC2 families with MMR-stable tumors.22, 23, 24, 25 The FCCTX subset is a major cause of hereditary colorectal cancer, although it remains a weakly defined and sparsely investigated subgroup of hereditary colorectal cancer. A better understanding of hereditary colorectal cancer may provide important clues to disease-predisposition and could contribute to molecular diagnostics, improved risk stratification, and targeted therapeutic strategies. These needs motivate our review of the clinical presentation, histopathologic features, and molecular mechanisms of FCCTX.
Clinical presentation, histopathologic features, genetic susceptibility, and recommendations for surveillance
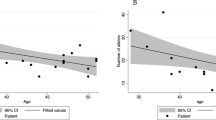
Registry data from Denmark, Finland, Italy, Australia, and from the Epicolon study suggest that 21–73% of the families that fulfil the AC1 and/or AC2 represent FCCTX16, 26, 27, 28 (Figure 1). Age at onset shows considerable interfamily as well as intrafamily variability with colorectal cancer diagnosed at a higher mean age (mean 57.3 years) in FCCTX than in Lynch syndrome (mean 49.7 years; Figure 1).16, 22, 29, 30, 31, 32 The FCCTX tumor spectrum is predominated by colorectal cancer and does, in contrast to Lynch syndrome, not show any increased risk of extracolonic cancers.16, 33, 34 Colorectal cancers linked to FCCTX are left sided in 70% of the cases. Synchronous as well as metachronous adenomas are frequent with a high adenoma/carcinoma ratio that may suggest a slower adenomacarcinoma progression rate than in Lynch syndrome.16, 21, 31, 33, 35, 36, 37, 38, 39

(a) Frequency of Amsterdam I/II families without disease-causing germline MMR mutations among different registers. (b) Mean age at diagnosis in Lynch Syndrome and FCCTX among the different registers. Source: Danish Hereditary NonPolyposis Register (Denmark), CCFR: Colorectal Cancer Family Register (Australia), Finnish Hereditary Register (Finland), Epicolon Study and Register of Hereditary Digestive Tumors at Foundation IRCCs National Cancer Institute of Milan (Italy).
Whereas colorectal cancers linked to Lynch syndrome are characterized by poorly differentiated tumors, mucinous differentiation, an expanding growth pattern, and abundant lymphocytic reactions (including tumor-infiltrating lymphocytes, peritumoral lymphocytes, and Crohn-like reactions), FCCTX tumors typically show a more ‘sporadic-like’ phenotype with medium high differentiation, glandular and infiltrative growth patterns, and frequent dirty necrosis (Figure 2).37,38 The lack of distinct histopathologic features makes identification of FCCTX-associated colorectal cancer challenging from a pathologist’s perspective and underscores the importance of obtaining a family history of cancer.

Hematoxylin and eosin-stained slides from colorectal cancers linked to FCCTX demonstrating (a) dirty necrosis and (b) infiltrative growth in the deep tumor margin. Dirty necrosis is characterized by the presence of large amounts of cell detritus and inflammatory cells within the glandular lumina, whereas the infiltrative growth pattern is characterized by the widespread dissemination of intraepithelial, mainly cytotoxic, T lymphocytes within the tumor tissue. Photo is courtesy of Professor Susanne Holck, Hvidovre Hospital, Denmark.
Surveillance programs in FCCTX are targeted at colorectal cancer and the mean age at onset of 60 years implies that surveillance colonoscopies are generally recommended with 3–5-year intervals, starting 5–10 years before the earliest age at onset in the family.3, 16, 19, 34, 40
A number of genome-wide association studies have addressed susceptibility loci in hereditary colorectal cancer, although not specifically linked to FCCTX. Candidate genetic variants have been reported in ‘CENPE (centromere protein E, 312 kDa)’ at 4q24–q25, ‘CDH18 (cadherin 18, type 2)’ at 5p14.3, ‘GALNT12 (UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 12, GalNAc-T12),’ ‘ZNF367 (zinc finger protein 367),’ ‘HABP4 (hyaluronan binding protein 4),’ and ‘GABBR2 (γ-aminobutyric acid (GABA) B receptor, 2)’ on chromosome 9, and ‘BMP4 (bone morphogenetic protein 4)’ at 8q23.3, ‘GREM1 (gremlin 1, DAN family BMP antagonist)’ and ‘KIF24 (kinesin family member 24)’ on chromosome 15, and ‘BCR (breakpoint cluster region)’ at 22q11.23, 41, 42, 43, 44, 45, 46 In addition, two microRNA genes, hsa-mir-491/KIAA1797 and hsa-mir-646/AK309218, have been associated, but additional data and independent validations are needed for application of these markers in clinical risk models.46 Current understanding suggests that the risk alleles identified are insufficient to independently account for FCCTX, but a combination of moderate and low-risk alleles could contribute to the familial aggregation.4, 39, 40, 41, 42, 47
Genomic and epigenetic profiles of FCCTX-associated colorectal cancer
Deranged DNA methylation is inversely associated with the MSI and the CpG island methylation phenotypes (CIMP) and has been demonstrated in 30–40% of colorectal cancers.48, 49, 50, 51, 52 Hypomethylation in long interspersed nucleotide element-1 (LINE-1) has been linked to familial CRC, including FCCTX, and is thought to interfere with chromosomal segregation and thereby enhance chromosomal instability (CIN).49, 51, 53, 54, 55, 56 Global DNA hypomethylation has been associated with poor prognosis, shorter survival, younger age of onset, and familial colorectal cancer risk.39, 51, 55, 56, 57, 58, 59 The predisposition to LINE-1 hypomethylation in FCCTX tumors gives an evidence for a link between distinct molecular signatures and phenotypes associated with specific epigenotypes.59 In hereditary colorectal cancer, relatively less is known about the patterns of specific histone modifications that also regulate gene expression through controlling chromatin conformation.52, 60 Recent studies have shown that mutation rates in colorectal cancer genomes are closely related to histone modification-directed chromatin organization and its upregulation is associated with a reduced patient survival,52, 61 suggesting a key role in colorectal cancer development.
The genomic profiles of FCCTX tumors show similarities to sporadic MMR-proficient colorectal cancer.42 Comparative genomic hybridization studies suggest that FCCTX tumors typically harbor 6–8 copy number alterations with recurrent gains of 7p, 7q, 8q, 13q, 20p, and 20q and losses of 17p, 18p, and 18q.62, 63 Gain of the 20q region has been specifically linked to FCCTX tumors and several candidate target genes such as ‘GNAS (GNAS complex locus),’ ‘AURKA (aurora kinase A),’ ‘SRC (v-src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog),’ ‘TOP1 (topoisomerase (DNA) I),’ ‘NELFCD (negative elongation factor complex member C/D),’ ‘ADRM1 (adhesion regulating molecule 1),’ ‘ASIP (agouti signaling protein),’ ‘CDH26 (cadherin 26),’ and ‘HNF1A (HNF1 homeobox A)’ reside herein (Figure 3).24, 62, 63 GNAS promotes proliferation through activation of the Wnt and ERK1/2 MAPK signaling pathways.64 Activating missense mutations in GNAS have been demonstrated in sporadic colorectal cancer, but the GNAS c.601G>T hot spot mutation could not be identified in a study of FCCTX tumors.63, 65 An overexpression of the chromosome-associated gene AURKA has been correlated to aneuploidy, invasion, and progression of colorectal adenoma to carcinoma.66 A candidate link also exists between gain of 20q and overexpression of ASIP, that could influence sensitivity to 5-fluorouracil.67 Loss of chromosome 18 is a common change in sporadic tumors as well as in FCCTX tumors and may be linked to downregulation of for example, ‘SMAD2 (SMAD family member 2),’ ‘SMAD4 (SMAD family member 4),’ ‘DCC (deleted in colorectal carcinoma),’ ‘SERPINB5 (serpin peptidase inhibitor, clade B (ovalbumin), member 5),’ and ‘BCL2 (B-cell CLL/lymphoma 2)’ (Figure 3).24, 25 Frequent (32%) genome-wide copy neutral loss of heterozygosity and a low frequency (14%) of chromosomal losses have been demonstrated in FCCTX tumors that could indicate involvement of yet unidentified DNA repair mechanisms.63, 68

Model illustrating identified genetic alterations that may promote tumorigenesis in FCCTX tumors. FCCTX are chromosomally instable with gains in chromosomal region 20q and complete loss of chromosome 18. These alterations may result in deregulation of genes involved in chromosomal segregation and genomic instability (AURKA), apoptosis (BCL2 and SERPINB5), proliferation (GNAS), growth inhibition, angiogenesis (PTGER1), and migration (SRC).
Mutations in several cancer-related genes such as ‘TP53 (tumor protein p53),’ ‘KRAS (Kirsten rat sarcoma viral oncogene homolog),’ ‘BRAF (v-raf murine sarcoma viral oncogene homolog B),’ APC, ‘MGMT (O-6-methylguanine-DNA methyltransferase),’ and ‘CTNNB1 (catenin (cadherin-associated protein), β1, 88 kDa)’ divide FCCTX tumors into two major groups; one-third of the tumors that are characterized by stable genotypes with few genetic changes retained membranous β-catenin expression and infrequent TP53 mutations, and two-thirds of the tumors with frequent loss of tumor suppressor gene loci such as APC, TP53, SMAD4, and DCC, somatic methylation of APC, KRAS, and MGMT, and nuclear translocation of β-catenin.22, 69 These genetic subsets have been suggested to differ in clinical presentation; genetically simple tumors predominantly develop in the proximal colon and develop at a lower (mean 54 years) age, whereas the genetically complex tumors more often develop in the distal colon and are diagnosed at a higher (mean 59 years) age.
Gene expression profiles and deranged signaling pathways
Gene expression studies in colorectal cancer have predominantly focused on differences between sporadic MMR-proficient and MMR-deficient tumors, whereas data on FCCTX tumors are scarce.70 MMR-proficient and MMR-deficient tumors show distinct profiles with 65–2070 significantly deregulated genes, including genes involved in growth factor receptors, transcription, cell cycle function, DNA repair, chromatin structure, drug metabolism, and chemoresistance.70, 71, 72, 73, 74, 75, 76 Gene expression data from FCCTX tumors suggest similarity to sporadic MMR-proficient colorectal cancers with upregulation of genes involved in peptidyl-amino acid modification, enzyme-linked receptor protein signaling, growth regulation, DNA repair pathways, vascular smooth muscle contraction, and G protein-coupled receptor signaling.25, 77 The limited data available regarding signaling pathways in FCCTX tumors indicate involvement of G protein-coupled signaling and candidate genes involved in proliferation and migration, for example, CDH26, SRC, and ASIP (located in chromosome 20q).24, 25 ‘PTGER1 (prostaglandin E receptor 1 (subtype EP1), 42 kDa)’ is activated by COX-2/PGE-2 signaling that is overexpressed in 90% of sporadic colon carcinomas and is induced by hypoxia.78, 79 Another target that has shown to directly upregulate COX-2 expression is the ‘HIST1H1A (histone cluster 1, H1a)’, whose overexpression had been significantly associated with a shorter colorectal cancer-specific and overall survival.80 Through a SRC-dependent pathway, PTGER1 activates the ‘ANGPTL4 (angiopoietin-like 4)’ protein that may promote colorectal carcinogenesis.79 Upregulation of this pathway may drive FCCTX tumor development during anaerobic conditions, and this is supported by frequent findings of dirty necrosis during histological evaluations and downregulation of the aerobic oxidative phosphorylation metabolism genes such as ‘ATP5L (ATP synthase, H+ transporting, mitochondrial Fo complex, subunit G),’ ‘ATP5A1 (ATP synthase, H+ transporting, mitochondrial F1 complex, α-subunit 1, cardiac muscle),’ ‘ATP5B (ATP synthase, H+ transporting, mitochondrial F1 complex, β-polypeptide),’ and ‘ATP5D (ATP synthase, H+ transporting, mitochondrial F1 complex, δ-subunit)’ (Figure 3).25, 37 Copy number gains and EGFR-mediated activation of SRC have been correlated to migration and invasion81 (Figure 3). The TGFβR-mediated growth inhibition is executed through SMAD2 and SMAD4, whereas BCL2 induces apoptosis. Hence, biallelic loss of these genes may give FCCTX tumors a growth advantage (Figure 3). The tumor suppressor ‘SERPINB5 (serpin peptidase inhibitor, clade B (ovalbumin), member 5)’ is activated by TP53 and induces apoptosis and inhibits migration and invasion of tumor cells.43 The current understanding of how gene expression profiles in FCCTX-associated tumors influence tumor development thus suggests increased proliferation, reduced apoptotic activity, and enhanced migration and invasion that could contribute to the poor prognosis suggested in this subgroup (Figure 3).
Conclusion
The FCCTX subset is challenging, not least as the clinical presentation and the histopathologic features mimic sporadic MMR-proficient tumors. The genetic causes are unknown, but candidate genes include, for example, CENPE, CDH18, GREM1, BCR, KIF24, GALNT12, ZNF367, HABP4, GABBR2, and BMP4. Differences in genomic and gene expression profiles do exist, for example gain of chromosome 20q, global hypomethylation, and upregulation of the G-protein coupled receptor signaling pathway. Taken together, current data suggest tumor development linked to inhibition of apoptosis, insensitivity to growth inhibitory signals, inhibition of angiogenesis, and increased migration and invasion that may be reflected in infiltrative growth patterns and frequent dirty necrosis. Extended and in-depth analyses of the FCCTX tumor genome, methylome, and proteome could in well-defined tumor series shed light on the basic mechanisms for potential application in refined diagnosis and targeted interventions.
References
Patel SG, Ahnen DJ . Familial colon cancer syndromes: an update of a rapidly evolving field. Curr Gastroenterol Rep 2012;14:428–438.
Pinol V, Castells A, Andreu M et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005;293:1986–1994.
Balmana J, Castells A, Cervantes A et al. Familial colorectal cancer risk: ESMO Clinical Practice Guidelines. Ann Oncol 2010;5:v78–v81.
Woods MO, Younghusband HB, Parfrey PS et al. The genetic basis of colorectal cancer in a population-based incident cohort with a high rate of familial disease. Gut 2010;59:1369–1377.
Center MM, Jemal A, Ward E . International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev 2009;18:1688–1694.
Castells A, Giardiello FM . Familial colorectal cancer screening: so close, so far. Gastroenterology 2013;144:492–494.
Groden J, Thliveris A, Samowitz W et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589–600.
Kinzler KW, Nilbert MC, Su LK et al. Identification of FAP locus genes from chromosome 5q21. Science 1991;253:661–665.
Nishisho I, Nakamura Y, Miyoshi Y et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991;253:665–669.
Sieber OM, Lipton L, Crabtree M et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. NEngl J Med 2003;348:791–799.
Amos CI, Keitheri-Cheteri MB, Sabripour M et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet 2004;41:327–333.
Howe JR, Roth S, Ringold JC et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998;280:1086–1088.
Sweet K, Willis J, Zhou XP et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA 2005;294:2465–2473.
Vasen HF, Mecklin JP, Khan PM et al. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991;34:424–425.
Vasen HF, Watson P, Mecklin JP et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999;116:1453–1456.
Lindor NM, Rabe K, Petersen GM et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005;293:1979–1985.
Trano G, Sjursen W, Wasmuth HH et al. Performance of clinical guidelines compared with molecular tumour screening methods in identifying possible Lynch syndrome among colorectal cancer patients: a Norwegian population-based study. Br J Cancer 2010;102:482–488.
Lynch HT, Lynch PM . Molecular screening for the Lynch syndrome–better than family history? N Engl J Med 2005;352:1920–1922.
Vasen HF, Moslein G, Alonso A et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007;44:353–362.
Rodriguez-Soler M, Perez-Carbonell L, Guarinos C et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 2013;44:926–932 e1; quiz e13-4.
Koh PK, Kalady M, Skacel M et al. Familial colorectal cancer type X: polyp burden and cancer risk stratification via a family history score. ANZ J Sur 2011;81:537–542.
Francisco I, Albuquerque C, Lage P et al. Familial colorectal cancer type X syndrome: two distinct molecular entities? Fam Cancer 2011;10:623–631.
Segui N, Pineda M, Navarro M et al. GALNT12 is not a major contributor of familial colorectal cancer type X. Hum Mutat 2014;35:50–52.
Therkildsen C, Jonsson G, Dominguez-Valentin M et al. Gain of chromosomal region 20q and loss of 18 discriminates between Lynch syndrome and familial colorectal cancer. Eur J Cancer 2013;49:1226–1235.
Dominguez-Valentin M, Therkildsen C, Veerla S et al. Distinct gene expression signatures in lynch syndrome and familial colorectal cancer type x. PLoS One 2013;8:e71755.
Moreira L, Balaguer F, Lindor N et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012;308:1555–1565.
Peltomaki P, Gao X, Mecklin JP . Genotype and phenotype in hereditary nonpolyposis colon cancer: a study of families with different vs shared predisposing mutations. Fam Cancer 2001;1:9–15.
Nieminen TT, Abdel-Rahman WM, Ristimaki A et al. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology 2011;141:e23–e26.
Boland CR, Goel A . Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073–2087 e3.
Jenkins MA, Southey MC, Giles GG et al. Rationale for, and approach to, studying modifiers of risk in persons with a genetic predisposition to colorectal cancer. Curr Oncol Rep 2007;9:202–207.
Lindor NM . Familial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndrome. Surg Oncol Clin N Am 2009;18:637–645.
de la Chapelle A, Hampel H . Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol 2010;28:3380–3387.
Lynch HT, Lynch JF, Attard TA . Diagnosis and management of hereditary colorectal cancer syndromes: Lynch syndrome as a model. CMAJ 2009;181:273–280.
Balmana J, Balaguer F, Cervantes A et al. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2013;6:vi73–vi80.
Mueller-Koch Y, Vogelsang H, Kopp R et al. Hereditary non-polyposis colorectal cancer: clinical and molecular evidence for a new entity of hereditary colorectal cancer. Gut 2005;54:1733–1740.
Jenkins MA, Hayashi S, O'Shea AM et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology 2007;133:48–56.
Klarskov L, Holck S, Bernstein I et al. Hereditary colorectal cancer diagnostics: morphological features of familial colorectal cancer type X versus Lynch syndrome. J Clin Pathol 2012;65:352–356.
Llor X, Pons E, Xicola RM et al. Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway. Clin Cancer Res 2005;11:7304–7310.
Ku CS, Cooper DN, Wu M et al. Gene discovery in familial cancer syndromes by exome sequencing: prospects for the elucidation of familial colorectal cancer type X. ModPathol 2012;25:1055–1068.
Bernstein IT, Myrhoj T . Surveillance for urinary tract cancer in Lynch syndrome. Fam Cancer 2013;12:279–284.
Houlston RS, Webb E, Broderick P et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet 2008;40:1426–1435.
Middeldorp A, Jagmohan-Changur SC, van der Klift HM et al. Comprehensive genetic analysis of seven large families with mismatch repair proficient colorectal cancer. Genes Chromosomes Cancer 2010;49:539–548.
Win AK, Hopper JL, Buchanan DD et al. Are the common genetic variants associated with colorectal cancer risk for DNA mismatch repair gene mutation carriers? Eur J Cancer 2013;49:1578–1587.
DeRycke MS, Gunawardena SR, Middha S et al. Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol Biomarkers Prev 2013;22:1239–1251.
Clarke E, Green RC, Green JS et al. Inherited deleterious variants in GALNT12 are associated with CRC susceptibility. Hum Mutat 2012;33:1056–1058.
Venkatachalam R, Verwiel ET, Kamping EJ et al. Identification of candidate predisposing copy number variants in familial and early-onset colorectal cancer patients. Int J Cancer 2011;129:1635–1642.
Kontham V, von Holst S, Lindblom A . Linkage analysis in familial non-Lynch syndrome colorectal cancer families from Sweden. PLoS One 2013;8:e83936.
Abuli A, Bessa X, Gonzalez JR et al. Susceptibility genetic variants associated with colorectal cancer risk correlate with cancer phenotype. Gastroenterology 2010;139:788–796 e1–6.
Ogino S, Kawasaki T, Nosho K et al. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer 2008;122:2767–2773.
Goel A, Nguyen TP, Leung HC et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer 2011;128:869–878.
Colussi D, Brandi G, Bazzoli F et al. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci 2013;14:16365–16385.
Bardhan K, Liu K . Epigenetics and colorectal cancer pathogenesis. Cancers (Basel) 2013;5:676–713.
Joensuu EI, Abdel-Rahman WM, Ollikainen M et al. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res 2008;68:4597–4605.
Fearon ER . Molecular genetics of colorectal cancer. Annu Rev Pathol 2011;6:479–507.
Antelo M, Balaguer F, Shia J et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS One 2012;7, e45357.
Pavicic W, Joensuu EI, Nieminen T et al. LINE-1 hypomethylation in familial and sporadic cancer. J Mol Med 2012;90:827–835.
Goel A, Xicola RM, Nguyen TP et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology 2010;138:1854–1862.
Ogino S, Nishihara R, Lochhead P et al. Prospective study of family history and colorectal cancer risk by tumor LINE-1 methylation level. J Natl Cancer Inst 2012;105:130–140.
Ogino S, Lochhead P, Chan AT et al. Molecular pathological epidemiology of epigenetics: emerging integrative science to analyze environment, host, and disease. Mod Pathol 2013;26:465–484.
Lao VV, Grady WM . Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol 2011;8:686–700.
Li F, Mao G, Tong D et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 2013;153:590–600.
Abdel-Rahman WM, Ollikainen M, Kariola R et al. Comprehensive characterization of HNPCC-related colorectal cancers reveals striking molecular features in families with no germline mismatch repair gene mutations. Oncogene 2005;24:1542–1551.
Middeldorp A, van Eijk R, Oosting J et al. Increased frequency of 20q gain and copy-neutral loss of heterozygosity in mismatch repair proficient familial colorectal carcinomas. Int J Cancer 2012;130:837–846.
Wilson CH, McIntyre RE, Arends MJ et al. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene 2010;29:4567–4575.
Sjoblom T, Jones S, Wood LD et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314:268–274.
Sillars-Hardebol AH, Carvalho B, Tijssen M et al. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut 2012;61:1568–1575.
Jin YT, Ying XX, Hu YH et al. aPKC inhibitors might be the sensitizer of chemotherapy and adoptive immunotherapy in the treatment of hASIPa-overexpressed breast cancer. Oncol Res 2008;17:59–68.
Lips EH, van Eijk R, de Graaf EJ et al. Integrating chromosomal aberrations and gene expression profiles to dissect rectal tumorigenesis. BMC Cancer 2008;8:314.
Sanchez-de-Abajo A, de la Hoya M, van Puijenbroek M et al. Molecular analysis of colorectal cancer tumors from patients with mismatch repair proficient hereditary nonpolyposis colorectal cancer suggests novel carcinogenic pathways. Clin Cancer Res 2007;13:5729–5735.
Danielsen SA, Cekaite L, Agesen TH et al. Phospholipase C isozymes are deregulated in colorectal cancer–insights gained from gene set enrichment analysis of the transcriptome. PLoS One 2011;6:e24419.
Watanabe T, Kobunai T, Toda E et al. Distal colorectal cancers with microsatellite instability (MSI) display distinct gene expression profiles that are different from proximal MSI cancers. Cancer Res 2006;66:9804–9808.
Bertucci F, Salas S, Eysteries S et al. Gene expression profiling of colon cancer by DNA microarrays and correlation with histoclinical parameters. Oncogene 2004;23:1377–1391.
Dunican DS, McWilliam P, Tighe O et al. Gene expression differences between the microsatellite instability (MIN) and chromosomal instability (CIN) phenotypes in colorectal cancer revealed by high-density cDNA array hybridization. Oncogene 2002;21:3253–3257.
Yuan Z, Sotsky Kent T, Weber TK . Differential expression of DOC-1 in microsatellite-unstable human colorectal cancer. Oncogene 2003;22:6304–6310.
Kim H, Nam SW, Rhee H et al. Different gene expression profiles between microsatellite instability-high and microsatellite stable colorectal carcinomas. Oncogene 2004;23:6218–6225.
Banerjea A, Ahmed S, Hands RE et al. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol Cancer 2004;3:21.
Lee WS, Seo G, Shin HJ et al. Identification of differentially expressed genes in microsatellite stable HNPCC and sporadic colon cancer. J Surg Res 2008;144:29–35.
Maekawa M, Sugano K, Sano H et al. Increased expression of cyclooxygenase-2 to -1 in human colorectal cancers and adenomas, but not in hyperplastic polyps. Jpn J Clin Oncol 1998;28:421–426.
Kim SH, Park YY, Kim SW et al. ANGPTL4 induction by prostaglandin E2 under hypoxic conditions promotes colorectal cancer progression. Cancer Res 2011;71:7010–7020.
Baba Y, Nosho K, Shima K et al. PTGER2 overexpression in colorectal cancer is associated with microsatellite instability, independent of CpG island methylator phenotype. Cancer Epidemiol Biomarkers Prev 2010;19:822–831.
Lau SK, Shields DJ, Murphy EA et al. EGFR-mediated carcinoma cell metastasis mediated by integrin αvβ5 depends onactivation of c-Src and cleavage of MUC1. PLoS One 2012;7:e36753.
Acknowledgements
We thank Dr Aung Ko Win from the Centre for Molecular, Environmental, Genetic and Analytic Epidemiology, Melbourne School of Population Health, The University of Melbourne, Australia, Dr Päivi Peltomäki from Department of Medical Genetics, Biomedicum Helsinki, University of Helsinki, Finland, Dr Castells from the Department of Gastroenterology, Hospital Clınic, IDIBAPS, CIBERehd, University of Barcelona, Spain, Dr Lucio Bertario from the Register of Hereditary Digestive Tumors at Foundation IRCCs National Cancer Institute of Milan, Italy, and the Danish HNPCC Register, Denmark, for providing information support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Dominguez-Valentin, M., Therkildsen, C., Da Silva, S. et al. Familial colorectal cancer type X: genetic profiles and phenotypic features. Mod Pathol 28, 30–36 (2015). https://doi.org/10.1038/modpathol.2014.49
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.49
Keywords
This article is cited by
-
New insights on familial colorectal cancer type X syndrome
Scientific Reports (2022)
-
Prevalence and spectrum of MLH1, MSH2, and MSH6 pathogenic germline variants in Pakistani colorectal cancer patients
Hereditary Cancer in Clinical Practice (2019)
-
Current clinical topics of Lynch syndrome
International Journal of Clinical Oncology (2019)
-
Differential expression of CK20, β-catenin, and MUC2/5AC/6 in Lynch syndrome and familial colorectal cancer type X
BMC Clinical Pathology (2017)
-
Familial Associations of Colorectal Cancer with Other Cancers
Scientific Reports (2017)
