
Abstract
A large body of evidence shows that p16INK4a overexpression predicts improved survival and increased radiosensitivity in HPV-mediated oropharyngeal squamous cell carcinomas.(OPSCC). Here we demonstrate that the presence of transcriptionally active HPV16 in oral cavity squamous cell carcinomas does not correlate with p16INK4a overexpression, enhanced local tumor immunity, or improved outcome. It is interesting that HPV-mediated oropharyngeal squamous cell carcinomas can be categorized as having a ‘nonaggressive’ invasion phenotype, whereas aggressive invasion phenotypes are more common in HPV-negative squamous cell carcinomas. We have developed primary cancer cell lines from resections with known pattern of invasion as determined by our validated risk model. Given that cell lines derived from HPV-mediated oropharyngeal squamous cell carcinomas are less invasive than their HPV-negative counterparts, we tested the hypothesis that viral oncoproteins E6, E7, and p16INK4a can affect tumor invasion. Here we demonstrate that p16INK4a overexpression in two cancer cell lines (UAB-3 and UAB-4), derived from oral cavity squamous cell carcinomas with the most aggressive invasive phenotype (worst pattern of invasion type 5 (WPOI-5)), dramatically decreases tumor invasiveness by altering expression of extracellular matrix remodeling genes. Pathway analysis integrating changes in RNA expression and kinase activities reveals different potential p16INK4a-sensitive pathways. Overexpressing p16INK4a in UAB-3 increases EGFR activity and increases MMP1 and MMP3 expression, possibly through STAT3 activation. Overexpressing p16INK4a in UAB-4 decreases PDGFR gene expression and reduces MMP1 and MMP3, possibly through STAT3 inactivation. Alternatively, ZAP70/Syk might increase MUC1 phosphorylation, leading to the observed decreased MMP1 expression.
Similar content being viewed by others

Main
Oral cavity squamous cell carcinoma (OCSCC) represents one of the most common malignancies of the head and neck. Over 20 000 cases and 5000 deaths in the United States were attributed to this disease in 2009.1 Currently, the Risk Model is the only validated prognosticator that predicts treatment failure for low-stage high-risk oral cavity squamous cell carcinoma, adjusted for confounders.2, 3, 4 We recently published a validation study on stage I/II patients showing a 42% positive predictive value with respect to locoregional recurrence associated with WPOI-5 worst pattern of invasion type 5 (WPOI-5, see below). Unfortunately, few treatment options are available after failure of first-line therapy of oral cancers.
Oral cavity squamous cell carcinomas typically arise after the accumulation of genetic insults caused by cumulative lifestyle ‘chemical’ exposures (ie, smoking and drinking), whereas many oropharyngeal squamous cell carcinomas (OPSCC) developing in never-smoker/drinkers are human papillomavirus mediated (HPV+). It is well established that HPV+ oropharyngeal squamous cell carcinomas are associated with improved outcome compared with HPV-negative counterparts.5, 6, 7, 8, 9, 10 A large body of published data show that p16INK4a overexpression, a downstream effect of high-risk HPV E7-Rb binding, predicts improved survival and increased radiosensitivity; however, the underlying mechanisms of this improved outcome remain controversial.11, 12, 13, 14, 15, 16, 17, 18 Furthermore, the reasons why oral cavity squamous cell carcinomas harboring transcriptionally active (ie, ‘driver infection’) HPV are not associated with improved outcomes remain to be elucidated. Here we demonstrate that transcriptionally active HPV in oral cavity squamous cell carcinoma does not correlate with either the overexpression of p16INK4a tumor suppressor protein or enhanced local tumor immunity. This is in direct contrast to HPV+ oropharyngeal squamous cell carcinomas that typically overexpress p16INK4a and manifest evidence of enhanced acquired tumor immunity.
Here we propose an additional mechanism for the improved outcome of HPV+ oropharyngeal squamous cell carcinomas, related to less aggressive tumor invasion. Histopathological invasion phenotype (termed WPOI) is one element of the Risk Model.2, 3, 4 Typically, HPV+ oropharyngeal squamous cell carcinomas can be categorized as having a ‘nonaggressive’ invasion phenotypes, whereas aggressive invasion phenotypes are more common in oral cavity squamous cell carcinomas. The aggressive invasion phenotypes can be subclassified into ‘WPOI-4’ and ‘WPOI-5,’ and the latter category is reserved for carcinomas with a dispersed pattern of invasion. Using tumor invasion assays, we demonstrated that cell lines derived from HPV-mediated cancers are less invasive than their HPV-negative counterparts. We then tested the hypothesis that p16INK4a overexpression in WPOI-5 cell lines suppresses invasion by modifying expression of extracellular matrix (ECM) remodeling genes. This was achieved using invasion assays, and assessing p16INK4a-responsive pathways by the nCounter Human Cancer reference kit (NanoString Technologies), kinomic profiling (PamStation®12, PamGene International), and integrated network modeling of p16INK4a altered gene expression and kinase activity with GeneGo MetaCore (Thompson Reuters).
Materials and methods
This study was approved by the Institutional Review Boards of the University of Alabama at Birmingham (UAB), Maurice and Gabriela Goldschleger School of Dental Medicine, Tel-Aviv University (Tel Aviv, Israel), and Department of Pathology, Continuum Health Partners, Beth Israel Medical Center (New York, NY). Tumors sites were classified as either oral cavity or oropharynx according to ICD-O-3 topography codes.19 Consecutive patients (total 156) diagnosed with primary oral cavity cancer were identified (UAB: 104 patients; Beth Israel: 28 patients; and Tel Aviv University: 24 patients). Pathological diagnoses were confirmed. Carcinomas were categorized with respect to WPOI, lymphocytic host response, and perineural invasion, and assigned an overall risk score; those with scores ≥3 points were classified as high risk.2, 3, 4 Electronic medical record data regarding patient age, gender, tumor site, self-reported race, AJCC TNM stage (seventh edition),19 and treatment (primary surgery, ±adjuvant treatment, primary chemotherapy/radiation) were abstracted. Histological criteria for exclusion included limited tumor depth of invasion (≤4 mm) and spindle cell variant of squamous cell carcinoma. All available data on smoking and alcohol intake were collected (ever smoked yes/no, pack-years, prior smoking, number of years quit smoking, any alcohol intake, ounces per week). Unfortunately, the smoking and alcohol data often lacked quantitative detail; therefore, they were analyzed only as categorical variables (yes/no). The study entry point was the date of primary treatment. Disease progression was classified as either locoregional recurrence or distant metastases, and entered as date of first event versus last known date without evidence of disease. Overall survival and disease-specific survival were assessed through the electronic medical records and Social Security Death Index (SSDI) as date of death versus last known date alive. Patients not found within the SSDI were coded as alive as of the last visit date.
To compare immune response with HPV between tumor organ sites, 20 HPV-positive and 21 HPV-negative oropharyngeal squamous cell carcinomas were selected randomly to compare CD4+ and CD8+ populations with HPV-positive and HPV-negative oral cavity squamous cell carcinomas. The nature of this patient cohort and the methods for HPV analysis have been recently reported.20
Sample Procurement
The glass pathology slides were used to morphologically guide tumor procurement from archival paraffin blocks and avoid sampling overlying squamous mucosa whenever present. Multiple 1 mm cores were procured and sterile technique was used to prevent contamination between specimens and great care was taken to avoid contaminating samples with adjacent squamous mucosa. The methodologies are detailed in our recent publications.20, 21
Assessment of Transcriptionally Active HPV16/18: RNA Extraction
Procured samples were deparaffinized, centrifuged, rehydrated, and centrifuged. Tissue was incubated in lysis buffer overnight to maximize digestion. RNA extraction was performed using TRIzol and chloroform. The upper aqueous RNA phase was removed, combined with 10 μl of 1 mg/ml glycogen (Roche Applied Science) and 1 ml of isopropanol, and stored overnight at −20 °C. The RNA was further purified by centrifuging and washing the pellets with 1 ml 70% ethanol (4 °C), at 12 000 r.p.m. for 30 min at 4 °C. The supernatant was removed and 70% ethanol washing step was repeated. The supernatant was again removed, and the RNA pellet was dried for 15 min at room temperature and resuspended in 30 μl RNase-free water. Any remaining residual genomic DNA was digested by DNase using the Turbo DNA-Free kit (Ambion) for 15 min at 37 °C, for a final volume of 50 μl. The extracted total RNA was stored at −80 °C. Total RNA concentrations were measured by NanoDrop Epoch Spectrophotometer System (BioTech, USA). Specimen preparation was performed in a room separate from the nucleotide extraction area, with regular decontamination (DNA AWAY and RNaseAWAY Surface Decontaminants, Molecular BioProducts) of all surfaces and pipettes.
Reverse Transcription, First Amplification, and Nested Real-Time Amplification
Type-specific nested primers were used to detect transcripts for HPV16 E6 and E7 and HPV18 E6 and E7, as previously described.20, 21 Primers to detect GAPDH transcripts were used as internal controls. The Bio-Rad iScript cDNA synthesis kit was used for reverse transcription of total RNA into cDNA. Only GAPDH-positive samples were studied for HPV. Samples were first screened for GAPDH expression. All samples that were GAPDH negative after first PCR were submitted to a second round of PCR. Cross-contamination was prevented by using separate sterile tips for each sample; for nested PCR, real-time PCR (RT-PCR) amplicon tubes were spun before the tubes were opened, and separate Eppendorf tube openers were used for transferring RT-PCR products to the nested PCR mix. The first amplifications for HPV16E6, HPV16E7, HPV18E6, and HPV18E7 were performed using 30 cycles of PCR; the 50 μl samples contained at least 50 ng of cDNA, 0.3 μM final concentration of each of the first primer set (∼250 bp) and 1.25 U of Terra PCR Direct Polymerase Mix (Clontech, USA). The following thermocycler conditions were used: denaturation at 94 °C for 2 min, 40 cycles of denaturation at 94 °C for 30 s, annealing at 57 °C for 60 s, extension at 72 °C for 2 min, and final extension at 72 °C for 10 min. The initial denaturation step in ‘hot start’ RT-PCR occasionally results in sample dropout due to evaporation; therefore, all reactions were run in triplicate. Samples were deemed positive if two of three reaction wells were positive. Every reaction included the following controls: HeLa (HPV18), SiHa (HPV16), formalin-fixed, paraffin-embedded (FFPE) HPV16+ oropharyngeal carcinoma, formalin-fixed, paraffin-embedded pancreatic carcinoma (HPV negative), and a blank sample without template. The second amplifications used RT-PCR, the Maxima SYBR Green/Fluorescein qPCR master Mix (Fermentas), and the Opticon2 detection system (Bio-Rad). This reaction contained 1:50 volumes of the first direct PCR reaction, 0.3 μM of the second set of nested primers, and the Maxima SYBR Green/Fluorescein qPCR Master Mix. These assays were performed in triplicate as follows: 10 min preincubation at 95 °C, 40 cycles of denaturation at 95 °C for 15 s, annealing at 58.6 °C for 30 s, extension at 72 °C for 30 s, and final extension at 72 °C for 5 min. The same positive and negative controls were used as described above. Amplicons were verified by Δ cycle threshold (ΔCT). False-positive results were avoided by (1) adhering to sterile technique, (2) requiring the detection of both type-concordant transcripts (E6 and E7) in order to designate specimens as positive, and (3) performing melting curve analysis on the amplicons from the final reaction. All negative control reactions were appropriate, including HeLa (HPV18) as a negative control for HPV16 RT-PCR and SiHa (HPV16) as a negative control for HPV18 RT-PCR.
Immunohistochemistry (IHC): p16INK4a
Epitope retrieval was performed with 0.02 M concentration of citrate buffer (pH 6.0) heated at 97 °C for 20 min. The E6H4 clone of p16INK4a antibody was used (MTM Laboratories, Westborough, MA, USA) in a 1:2 dilution. Positive controls consisted of HPV16+ tonsillar carcinoma with strong diffuse nuclear and cytoplasmic p16INK4a expression. The negative control slides consisted of tissue sections processed without primary antibody. IHC was performed with a semi-automated immunostainer (Thermo Scientific, Labvision 720, Fremont, CA, USA) and the UltraVision LP polymer system and diaminobenzidine tetrachloride (DAB) chromagen was used to visualize the antibody–antigen complex; slides were counterstained with hematoxylin. The p16INK4a expression was categorized for staining intensity (scale 0 to 4) as well as percent staining distribution for both nuclei and cytoplasm. of both squamoid and mucinous tumor elements.Tumors were classified as p16INK4a positive if they expressed both nuclear and cytoplasmic p16INK4a with ≥+2 intensity and ≥75% distribution; any amount of staining less than this cutoff was classified as negative.22 The slides were examined blinded to HPV status.
CD4 and CD8
To compare local immune response with HPV status, 46 oral cancers were randomly selected and compared with resection specimens from 41 oropharyngeal cancers with known transcriptionally active HPV16/18 status.29 H&E slides were reviewed and one section with the strongest cancer-associated lymphocytic response was selected from each case for study. Tissue sections were pretreated with heat-induced epitope retrieval with 10 mM EDTA, pH 8.0, for 20 min at 98 °C followed by cooling at room temperature for 20 min. Endogenous peroxidase activity was blocked by incubation in the hydrogen peroxide block (Thermo Scientific). After washing in Tris-buffered saline with 0.1% Tween-20, a nonserum protein block was applied for 15 min at room temperature (Ultra V Block; Thermo Scientific/LabVision). For detection of CD4 and CD8, we used the primary antibodies with a ready-to-use MultiVision polymer cocktail consisting of anti-mouse/horseradish peroxidase and anti-rabbit/alkaline phosphatase (Thermo Scientific MV-2001R7). Whole slide scanning was performed (BioImagene); ten × 400 fields were selected from the regions of densest tumor-infiltrating-lymphocytes (TILS), and densest immune response at the tumor interface. CD4 and CD8 positive cells were quantified as percent cells per × 400 field.
Immunofluorescence of FOXP3
To better characterize the nature of the CD4 cells, Tregs were quantified by double staining immunofluorescence with FOXP3/CD4 antibodies (Thermo Scientific). A total of 20 oropharyngeal squamous cell carcinomas (9 HPV-positive and 11 HPV negative) were randomly selected for study from among the 41 cases above. After rehydration, heat-mediated citrate buffer antigen retrieval was performed. Slides were incubated overnight with 5 μg/ml FOXP3 antibody (Abcam, ab4728), washed, and stained with Alexa Fluor 594 goat anti-rabbit antibody (Life Technologies, 45 min, 1:500 dilution). After extensive washing, the second overnight incubation with CD4 antibodies (Thermo Scientific, MS-1528-S) was performed. Slides were washed and incubated with goat anti-mouse secondary antibody conjugated to Alexa 488 (Life Technologies) for 45 min in a 1:500 dilution. Diamidino-2-phenylindole hydrochloride (DAPI) staining was performed to localize nuclei. Negative control tissues were treated in the same way, but incubated only with secondary Alexa Fluor-labeled antibody. Slides were washed and mounted in Vectashield medium for fluorescence (Vector Labs, CA, USA). Double-stained FOX3P+/CD4+ (Treg) cells appeared as yellow fluorescence, and were quantified by cell counting in the ten × 40 fields with the greatest number of cells.
UAB Cell Lines and Transfection
We have developed unique squamous carcinoma cell lines from primary head and neck cancer resections with known tumor ‘WPOI’ (Table 1 and Figure 1). The cell lines were double sorted using PE-A-conjugated Epcam/CD326 antibody (epithelial cell adhesion molecule), and their epithelial nature was confirmed on cytospin preparations that were fixed and permeabilized in cold acetone and stained with keratin cocktail antibodies (not shown). HPV status was confirmed by PCR with GP5+/6+ primers plus reverse transcription and PCR with HPV16/HPV18-type specific E6 and E7 primers.

Worst patterns of invasion (WPOI). (a) Tongue squamous cell carcinoma, WPOI-5, × 2 magnification. Satellite (S) is >3 mm away from the next closest wave of tumor satellites (T). Note the interspersed large muscle bundles in region T, typical of a WPOI-5 squamous cell carcinoma. (b) Another WPOI-5 tumor with strand-like dispersed tumor islands, the requisite distance (≥1 mm) is not illustrated in this photomicrographic field. (c) In contrast, WPOI-3 is a nondispersed, nonaggressive pattern of invasion. (d, e) Another way of visualizing WPOI-5 and WPOI-3 in the same cancers as (b) and (c). White indicates carcinoma and black indicates stroma. Images generated by IHC for keratin, whole slide digital scanning, and processing with ImageJ program. (f) HPV-mediated tonsil carcinoma. At low-power magnification, the nonkeratinizing squamous cell carcinoma is barely discernible from the tonsillar lymphoid tissue. (g) Same tumor, immunohistochemistry for p16. The pattern of p16 expression distinguishes tumor islands (brown) from lymphoid tissue (blue). Histological examination of the tumor interface at high-power allows for the classification of this tumor as WPOI-3.
To examine the impact of HPV viral oncoproteins and p16INK4a on tumor invasion, HPV-negative cell lines were Lipofectamine transfected with plasmid vectors containing cloned HPV16E6, HPV16E7, and p16INK4a. pCMV-Sport2 vector was used for HPV16E6 and HPV16E7 transfection. pCMV6-CDKN2A plasmid was used for p16INK4a transfection.
To determine whether the impact of overexpressing p16INK4a is specific to p16INK4a, and reversible, cells were co-transfected with p16INK4a expression vector and silencing hairpin RNA. RNA silencing was achieved by lentiviral plasmid transfection (pLenti X2 Hygro/sHp16 (w192-1) vector (Addgene)). Lentiviral plasmid transfection with sH scramble RNA was used as a negative control. SCC-90, an HPV16-positive cell line from squamous cell carcinoma of the tongue base, was transfected with either sH scramble or sH p16INK4a as control. Transfection efficiency was estimated by performing either immunofluorescence or IHC for the relevant protein on cell line cytospin preparations. All invasion experiments were performed within 48 h of transfection.
Transwell Invasion Assay
Invasion was measured using BD BioCoat Matrigel Invasion Chambers (BD Biosciences, San Jose, CA, USA) with 8 mm diameter, matrigel-coated polycarbonate membrane (8 μm pore size). A total of 105 tumor cells were seeded into the upper chamber of the well. Chemotaxis was achieved with 100 pg/ml of TGF-β1 for 36 h. Then, the cells on the upper surface of the filter were wiped off using a cotton swab, and the cells that had invaded the underside of the filter were fixed, stained with DiffQuick, and counted under bright-field microscopy at × 10 magnification in five random fields of view. Each condition was assayed in triplicate.
FlouroBlok Invasion System
The FlouroBlok invasion system was also used to study the invasiveness of the tumor cells. 3T3 cells, the mouse fibroblast cell line (ATCC) of low invasivity, were used for control. The FluoroBlok invasion system consists of a BD Falcon™ FluoroBlok 24-multiwell insert plate with an 8 μm pore size and polyethylene terephthalate membrane that has been uniformly coated with BD Matrigel Basement Membrane Matrix. 105 tumor cells were seeded into upper chamber of the well. The bottom chambers contained DMEM with 10% fetal calf serum as a chemoattractant. The plates were incubated at 37 °C, 5% CO2 atmosphere for 48 h. Following incubation, the medium was removed and inserts were transferred into a second 24-well plate containing 0.5 ml/well of 4 mg/ml Calcein AM in Hanks’ buffered salt solution (HBSS). Plates were incubated for 1 h at 37 °C, 5% CO2. Invasive cells were quantified as relative fluorescence units and read in a fluorescence plate reader Synergy 2 (BioTek) at 485/530 nm. Each condition was studied with three to five replicates. Sham transfection with empty vector (EV) served as negative controls with transfection experiments. Multiple invasion assays were performed comparing replicates of p16INK4aUAB-3, p16INK4aUAB-4, sh-RNA-p16INK4aUAB-3, sh-RNAp16INK4aUAB-4, double transfectants of p16INK4a plus sh-RNA-p16INK4a, plus appropriate controls.
Cell Proliferation Assay
Cells were seeded in 96-well plates (2 × 103) and allowed to attach overnight. Cell proliferation was measured at 0, 24, 48, and 72 h using the CellTiter 96® AQueous One Solution Cell Proliferation Assay CellTiter 96 Aqueous kit (Promega). Assays were performed by adding 20 μl of the CellTiter 96 AQueous One Solution Reagent to 100 μl of cell culture, incubating for 1 h, and then recording absorbance at 490 nm with a 96-well plate reader (Epoch, BioTek). The concentration of the reaction product, formazan, was directly proportional to the number of living cells in culture product.
RNA Expression Analysis
The nCounter Human Cancer reference kit (NanoString Technologies) was used to study changes in gene expression after p16INK4a overexpression or silencing. The Human Cancer reference kit queries a broad set of 230 genes relevant to cancer-related processes including invasion, cell cycle, proliferation, apoptosis, etc. Total RNA at 100 ng was analyzed from each of the following: empty vector control-UAB-3, p16INK4aUAB-3, sH RNA p16INK4aUAB-3, empty vector control-UAB-4, p16INK4aUAB-4, and sH RNA p16INK4aUAB-4. Gene expression data were normalized to six internal housekeeping genes. Changes in gene expression were analyzed as (p16INK4a overexpression)/(empty vector transfectants) and (sH RNA p16INK4a silencing)/(empty vector transfectants). The data were examined for the impact of p16INK4a overexpression alone or compared with the impact of p16INK4a silencing. The cutoff for evaluation was taken as a ratio of ≤0.66 or ≥1.5. If the ratios for overexpression and silencing were significant and reciprocal (eg, decreased gene expression with p16INK4a overexpression plus increased gene expression with p16INK4a silencing), then that gene is considered potentially interesting.
Western Blot
Tumor cell lines were lysed in the cold lysis buffer (Pierce) containing protease and phosphatase inhibitors (Thermo Scientific). Equal amounts of protein (40 μg) were separated on 10% polyacrylamide gel. Proteins were then transferred from the gel to nitrocellulose membrane (Amersham, GE Healthcare) using the Bio-Rad Laboratories Mini Protein 3 system. The membranes were treated with blocking buffer (Li-COR) for 1 h at room temperature and the primary antibodies diluted in the blocking reagent were applied. Antibodies were diluted according to the manufacturers’ recommendation (p16 and MMP1, Millipore and β-actin, Genetex). After overnight incubation at 4 °C, membranes were washed extensively with TBST and incubated for 1 h at room temperature with a secondary antibody Goat anti-Rabbit IRDye 680 or Goat anti-mouse IRDye 800 (1:10 000 dilution; Li-COR) in blocking buffer. Finally, membranes were washed extensively with TBST and imaged using Odyssey imaging system.
Kinomics Analysis
Kinomic profiling of p16INK4aUAB-3, empty vector control-UAB-3, p16INK4aUAB-4, and empty vector control-UAB-3 lysates was performed utilizing a high-throughput phosphorylatable-peptide microarray platform, the PamStation12 (PamGene, Hertogenbosch, The Netherlands), within the UAB Kinome Core (www.kinomecore.com). This fluorescent-detection system measures the ability of the active kinases in a specimen to phosphorylate specific peptides, imprinted on multiplex arrays, in real time with kinetic evaluation allowing for kinomic profiling of cells and tissue. Monolayer cultures were lysed in M-PER lysis buffer with protease and phosphatase inhibitors at 4 °C for 30 min. Then, 15 μg of protein was combined with kinase buffer, ATP, and fluorescently labeled anti-PY20 antibodies per UAB Kinome Core standard operating procedure and then loaded in triplicate into each well of the tyrosine (PTK) PamChips®. The samples were run on the PamStation12 kinomics workstation using the standard PTK PamChip protocol using Evolve12 Software v. 1.5 (PamGene). Image analysis and quantitation including heatmaps and unsupervised hierarchical clustering were performed using BioNavigator v. 6.0 (PamGene). Raw signal intensity data per each of the 144 spots were captured over multiple 50 ms exposures sequentially as ‘prewash’ kinetic values, and then over multiple exposure times (10, 20, 50, 100, and 200 ms) as ‘postwash’ values. These postwash values were converted to slopes of intensity by exposure time, and slopes were multiplied by 100 and log2 transformed for visualization and are labeled ‘postwash’ values. Significantly altered peptides (P<0.05, by paired Student’s t-test), generated from ±p16INK4a overexpressing lysates with acceptable phosphorylation curves, were plotted as prewash values. Significantly altered peptides were cross-compared for recurrent upstream kinases as listed per residue in Kinexus kinase predictor (www.phosphonet.ca), and scored according to percent occurrence. Upstream kinases and altered genes were uploaded to GeneGo MetaCore (portal.genego.com, a Thomson Reuters Company), a networking tool with curated biological interactions, for network analysis.
Statistical Analysis
Associations between HPV16+ status, p16INK4a status, pathological T stage, lymph node status (positive versus negative), cigarette and alcohol intake, high-risk status, disease progression, and disease-specific mortality were assessed by Fisher’s exact test or χ2 test; all tests were two tailed unless stated, with a probability value of <0.05 considered statistically significant. Univariate Kaplan–Meier analysis was performed to assess time to disease progression and overall survival for HPV status, race, and other demographics. Multivariate modeling was performed for both HPV16 and p16INK4a, adjusted for age (continuous variable), gender, smoking, alcohol, high risk, race, and center. Paired t-tests were performed comparing for mean cell counts between CD8 tumor-infiltrating lymphocytes, CD4 at the interface, and Foxp3+/CD4+ cells, and with altered peptides in the kinomics analysis; significance was set as P<0.05.
Results
p16 Overexpression, HPV16, and Outcome
With respect to HPV, a total of 155 and 120 patients were studied for HPV16 and HPV18, respectively; 112 patients were studied for p16. Overexpression of p16INK4a is uncommon in oral cavity squamous cell carcinoma (17/112 or 15.2%) and did not correlate with HPV16 status. HPV16 and HPV18 were detected in 22.6% (35/155) and 11% (13/120) patients, respectively (Figure 2). Table 2 presents the frequency distribution of p16INK4a overexpression and HPV16 by clinicopathologic features, expressed as actual patient numbers. Multivariate modeling demonstrated a protective effect with p16INK4a overexpression and disease progression, when adjusted for confounders (Table 3, bold font: P<0.05, hazard ratio 0.11, 95% CI 0.02–0.63). Kaplan–Meier analysis and multivariable modeling confirmed the lack of association with disease progression or disease-specific mortality (not shown).

Oral squamous cell carcinoma, transcriptionally active HPV, and p16. (a) Reverse transcription, PCR with primers for HPV16E6 (left) and HPV16E7 (right). Lanes 1 and 2: HPV16+ oral cavity squamous cell carcinoma; lane 3: SiHa (HPV16+); lane 4: HeLa (HPV18+); lane 5: formalin-fixed, paraffin-embedded negative tissue control. (b) Rare p16+ WPOI-5 oral cavity squamous cell carcinoma, × 4, × 10, immunohistochemistry. (c) p16-negative oral cavity squamous cell carcinoma, × 4, × 10, immunohistochemistry.
Local Adaptive Immunity and HPV Status
Figure 3 compares local adaptive immunity with HPV status in a subset of 46 patients with oral cavity squamous cell carcinoma, and 41 patients with oropharyngeal squamous cell carcinoma, respectively. No association is seen with HPV+ and CD4+ or CD8+ cells for oral cavity squamous cell carcinomas. In contrast, HPV+ oropharyngeal squamous cell carcinomas are significantly more likely to be infiltrated with more intratumoral cytotoxic CD8+ cells and tumor interface CD4+ cells than HPV− counterparts. The nature of the CD4+ cells, studied further in a subgroup of oropharyngeal squamous cell carcinomas, demonstrated a significant and inverse relationship between Tregs and CD8+ cells. HPV+ oropharyngeal squamous cell carcinomas harbored low Tregs (Foxp3+/CD4+) and high CD8+ tumor-infiltrating lymphocytes compared with HPV− oropharyngeal squamous cell carcinomas that harbored high Tregs and low CD8+ tumor-infiltrating lymphocytes (Figure 4, P=0.0007, paired two-tailed t-test).

Local adaptive immunity to squamous carcinoma by HPV status and anatomic subsite. Brown cells: CD4+, blue cells: CD8+. Counterstain enhanced for purposes of demonstrating carcinomas. (a) HPV16+ oral cavity squamous cell carcinoma ( × 10 magnification). (b) Limited immune response in this HPV16+ oral cavity squamous cell carcinoma ( × 200 magnification). (c) HPV-negative oral cavity squamous cell carcinoma ( × 10 magnification). Note CD4+ cells (brown) predominantly at interface. (d) Note intratumoral cytotoxic CD8+ cells (blue). No significant relationship is found between immune response and HPV status for oral cavity squamous cell carcinoma ( × 200 magnification). (e) HPV+ tonsil carcinoma ( × 10 magnification). Quantification of CD4+/CD8+ cells confirm that mean cytotoxic tumor-infiltrating lymphocytes and interface CD4 cells are significantly greater in HPV+ oropharyngeal squamous cell carcinomas as compared with HPV-negative counterparts. (f) HPV+ tonsil carcinoma ( × 200 magnification). (g) HPV-negative tonsil carcinoma ( × 10 magnification). (i) HPV-negative tonsil carcinoma ( × 200 magnification).

Tregs in oropharyngeal carcinomas by HPV status. To better characterize the nature of the CD4 cells that were significantly elevated in HPV+ oropharyngeal squamous cell carcinomas, Tregs were quantified by immunofluorescence as FOXP3+/CD4+ in a subgroup of 20 oropharyngeal squamous cell carcinomas (9 HPV+ and 11 HPV negative). Foxp3+/CD4+ cells (right panel, yellow fluorescence, arrows) represent Tregs. A significant inverse relationship is seen between higher CD8+ and lower Treg populations in HPV+ oropharyngeal squamous cell carcinomas, compared with lower CD8+ and higher Treg populations in HPV-negative counterparts (paired two-tailed test P=0.0007).
Taken together, the above data suggest that the lack of a protective effect for patients with oral cavity squamous cell carcinoma harboring HPV16 can also be ascribed to: (1) the lack of association between HPV16 and p16INK4a overexpression, and (2) the absence of increased local adaptive tumor immunity associated with HPV infection. The multivariate modeling data also confirm a significant survival advantage with respect to disease progression for p16INK4a overexpression.
Tumor WPOI and Cell Line Invasion
The p16INK4a was overexpressed in 2/39 cancers classified as WPOI-5 (Figure 2), and 11/47 cancers classified as WPOI-4. The overexpression of p16INK4a was significantly less likely to coincide with WPOI-5 compared with WPOI-4 (P=0.032, Fisher’s exact test, two tailed). Multiple pilot transwell invasion assays and subsequent FluoroBlok invasion assays demonstrated that cell lines derived from carcinomas with WPOI-4 and WPOI-5 (UAB-3, UAB-4, and UAB-5; Table 1) demonstrate greater invasiveness compared with cell lines derived from carcinomas with WPOI-3 (UAB-1 and UAB-6 (both HPV16+), as demonstrated in Figure 5).

Invasiveness of tumor cell lines by HPV status and worst patterns of invasion (WPOI). Transwell invasion assays (a) and FluoroBlok invasion assays (b) demonstrate that HPV16+ UAB-1 and UAB-6 are significantly less invasive than UAB-3 and UAB-4, both of which are HPV negative and derived from WPOI-5 oral cavity squamous cell carcinoma.
HPV-negative cell lines (UAB-4 and UAB-5) co-transfected with HPV16E6/E7 (transfection efficiency at 50%) were significantly less invasive than pilot transwell invasion studies (Figure 6). Subsequent invasion experiments with UAB-3 and UAB-4 (both WPOI-5) transfected with E6, E7, or E6E7 were assessed by the FluoroBlok invasion assay. Multiple experiments confirmed that sham transfection with empty vector (EV) controls did not affect invasion. E7-UAB-3 demonstrated 10.7% decreased invasion (P=0.0016) (not shown).

Impact of E6/E7 on cell line invasion. (a) Confirmation of E6/E7 cloning into plasmid vector: nontransfected and E6/E7-transfected plasmid vectors, lane 1 versus lanes 2 and 3, respectively. PCR was performed using E6 primers and E7 primers, lanes 1 and 2 versus lane 3, respectively. (b) Confirmation of HPV transfected in cell lines: reverse transcription and PCR was performed using HPV16 E6 primers on Sham-UAB-4, E6E7-UAB-4, Sham-UAB-5, and E6E7-UAB-5, with SiHa (HPV16+) as positive control. (c) Transwell invasion assay: tumor cells that invade through the 8 μm round pores (circles, *) are visually after Diff-Quik counterstain (black arrows). Left panel: Sham-UAB-5; right panel: transfected E6E7-UAB-5. Fewer invasive cells are seen in UAB-5 after E6E7 co-transfection. WPOI, worst pattern of invasion; Δ, change; 95% CI, 95% confidence interval.
The cell lines UAB-3 and UAB-4 were confirmed as p16INK4a nonexpressors, and then phenotypic alterations associated with p16INK4a overexpression were demonstrated in these cell lines (transfection efficiency at 50%). Histologically, p16INK4a transfectants appear more mature with greater cytoplasm and lower nuclear/cytoplasmic ratio compared with sham transfectants. Overexpression of p16INK4a significantly decreased tumor invasion for all experiments (Figure 7).

Overexpressing p16INK4a increases maturation. Sham-transfected-UAB-3 (a, b) and p16INK4a-UAB-3 (c, d) cytospin preparations. (a, c) Stained with hematoxylin and eosin (H&E), × 20 original magnification. (b, d) Immunohistochemistry (IHC) for p16, × 40 original magnification. p16INK4a-UAB-3 demonstrates increased maturation with decreased nuclear to cytoplasmic ratio. Sham-transfected UAB-3 is p16 negative, and p16INK4a-UAB-3 is confirmed as an p16INK4a expressor. Successful p16INK4a transfection was also confirmed by western blot analysis (lane 1, Sham-transfected-UAB-3; lane 2, p16INK4a-UAB-3).
The efficiency of p16INK4a silencing was >95% (Figure 8). The p16INK4a silencing alone did not affect invasion. Figure 9 demonstrates that p16INK4a overexpression significantly decreased invasion by 68.1% and 61.0% for UAB-3 and UAB-4, respectively. The impact of p16INK4a overexpression on invasion was abrogated by co-transfection with p16INK4a and silencing hairpin p16INK4a RNA (Figure 9a). The rate of proliferation of p16INK4a-UAB-3 and p16INK4a-UAB-4 was decreased 24% and 14%, respectively, at 48 h, corresponding to the same time point as the invasion experiments (Figure 9c and d).

Silencing p16INK4a. (a) Cytospin preparation of SCC-90 (HPV16+), H&E. (b) Transfection with sH scramble RNA, IHC for p16INK4a. (c) Transfection with sH p16INK4a RNA, IHC for p16INK4a.

Overexpressing p16INK4a decreases invasion. (a) FluoroBlok invasion assays for UAB-3 and UAB-4, comparing empty vector (EV) transfection, p16INK4atransfection, and double transfection with p16INK4a, and p16INK4a silencing hairpin RNA. The p16INK4a overexpression decreased invasion by 68.1% for UAB-3 and 61.0% for UAB-4. This effect was abrogated by co-transfection with p16 and sh p16RNA (y axis=relative fluorescent units, FluoroBlok invasion assay, 48 h, EV=empty vector). (b) Undersurface of the FluoroBlok membranes from these experiments. The cells that invade through the 8 μm pores are labeled with calcein (fluprexon). (A) UAB-3 control, (B) p16INK4aUAB-3, (C) UAB-4 control, and (D) p16INK4aUAB-4. The numbers of invading tumor cells are significantly decreased with p16INK4a overexpression. (c, d) Tumor proliferation assays for UAB-3 (3) and UAB-4 (4). The p16INK4a overexpression also decreased tumor proliferation by 24% for p16INK4aUAB-3 and 14% for p16INK4aUAB-4 at day 2, corresponding to the same time point as the invasion experiments. The p16INK4a overexpression decreased tumor invasion in excess of the degree of decreased tumor proliferation.
Expression Array Analysis
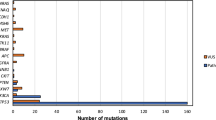
For both cell lines, overexpressing p16INK4a resulted in significant downregulation of multiple genes, whereas fewer genes were upregulated. Interestingly, different gene expression response patterns were observed for UAB-3 and UAB-4, even though the magnitude of dampened invasion with p16INK4a overexpression was similar for both cell lines. For instance, p16INK4a overexpression silenced MMP1 in UAB-4, but upregulated MMP1 in UAB-3. Similarly, MMP3 demonstrated opposite response patterns to p16INK4a overexpression for both cell lines. The heat map analysis (Figure 10d) imposes an additional level of stringency; that is, the impact of p16INK4a overexpression and silencing are reciprocal. Three of the seven genes of interest that are common to both cell lines are directly involved in extracellular matrix remodeling: TIMP3, COL1A1, and PDGFRA.

Differences in p16INK4a-sensitive invasion pathways. (a) Fold decreased gene expression with p16INK4a overexpression for UAB-3 (blue) and UAB-4 (red). MMP1 and COL1A1 were decreased more than 40-fold in UAB-4. (b) Decreased MMP1 protein was confirmed by western blot (empty vector-UAB-4 versus p16INK4a-UAB-4). (c) Fold increased gene expression with p16INK4a overexpression for UAB-3 (blue) and UAB-4 (red). MMP3 and MMP1 expression were significantly increased for UAB-3, but decreased for UAB-4 under the same conditions. (d) For a given gene, if (sH RNA transfectants)/(empty vector transfectants) ≤0.66 or ≥1.5, and (p16INK4a transfectants)/(empty vector transfectants) ≤0.66 or ≥1.5, with a reciprocal relationship, then that gene is considered potentially interesting. For both cell lines, overexpressing p16INK4a resulted in significant downregulation of multiple genes, but upregulation of fewer genes. Genes that are significantly downregulated by p16INK4a and common to both cell lines are highlighted in yellow. This includes three genes involved in extracellular matrix remodeling: TIMP3, COL1A1, and PDGFRA.
Kinomics Analysis
Overexpression of p16INK4a in UAB-3 and UAB-4 increased overall general kinase activity compared with controls (preponderance of red vs blue cells in p16-labeled columns); unsupervised hierarchical clustering of the full tyrosine kinome demonstrated that sample replicates clustered primarily by cell line (Figure 11). The differences in kinase activities altered by p16INK4a overexpression between UAB-3 and UAB-4 predicted different upstream kinases (Figure 12), reflecting differences seen between the two in terms of the gene expression array data. Whereas UAB-3 demonstrated increased EGFR-type signaling with p16INK4a overexpression, UAB-4 cells demonstrated increased ZAP70-type signaling.

The p16INK4a-sensitive kinomic signatures. Unsupervised hierarchical clustering of full tyrosine kinome (144 peptides, one peptide per row, y axis; one cell line replicate per column, x axis). Red indicates increased and blue indicates decreased relative signal deviation from experimental mean (log2 transformed slope × 100).

The p16INK4a-sensitive peptide phosphorylation and kinase activities. Kinase activity (amount of phosphorylation) for significant source peptides for UAB-3 (top) and UAB-4 (bottom) shown with (a) kinetic curves for altered phosphopeptides (increased phosphorylation denoted by height on the x axis, and time on the y axis per cell line replicate) and (b) increased upstream kinases from scores derived from the Kinexus database for UAB-3 (top) and UAB-4 (bottom). Control is green, and p16 overexpression samples are light blue. A full color version of this figure is available at the Modern Pathology journal online.
Network mapping of the predicted upstream kinases with the gene expression data (limited to genes with ≥10-fold change) further identified different pathways that could explain the observed differences between the UAB-3 and UAB-4 with respect to p16INK4a-sensitive changes in metalloproteinase expression. For UAB-3, the most straightforward pathway linking the kinase and gene expression changes is EGFR stimulation of STAT3 that can then upregulate MMP1 and MMP3 (listed as stromelysin 1 in the network maps, Figure 13b). For UAB-4, two potential pathways can lead to decreased MMP1 expression (Figure 13d). First, the observed decreased PDGFR expression could lead to decreased STAT3 activation, thereby reducing MMP1 and MMP3 expression and activity. Alternatively, ZAP70/Syk phosphorylation of MUC1 could lead to decreased expression of MMP1.

Networking p16INK4a-sensitive genes with peptide phosphorylation and kinase activities. UAB-3 altered genes (a) and network modeling combined with altered upstream kinases (b). UAB-4 altered genes (c) and network modeling combined with altered upstream kinases (d). Actual data in round blue ‘nodes’, annotated with either pink circles (increased kinase), dark red circles (increased RNA), or dark blue circles (decreased RNA). Direction of interactions is denoted by arrowheads; positive interactions are green and negative interactions are red. A full color version of this figure is available at the Modern Pathology journal online.
Discussion
Genetic profiling reveals significant differences between HPV-negative and HPV-mediated head and neck squamous cell carcinoma. Chromosomal losses at 3p11.2–26.3, 5q11.2–35.2, 9p24–9p21.1 (region of CDKN2A or p16INK4a), and 17p (region of p53) and gains or amplifications at 11q12.1–11q13.4 (region of CCND1 (cyclin D1)) are more common in HPV-negative head and neck squamous cell carcinoma than HPV+ counterparts.23, 24, 25 This supports the idea that different pathways of carcinogenesis affect treatment response and outcomes. The high prevalence of wild-type tumor suppressor p53, Rb, and p16INK4a in HPV-mediated oropharyngeal squamous cell carcinoma represents one mechanism for the observed improved outcomes.
Overexpression of p16INK4a is a well-accepted surrogate biomarker of transcriptionally active HPV in the context of the cervix and oropharynx. High-risk (HR) HPV E7 oncoprotein binds Rb, targeting it for degradation; the oncogenic stress of Rb functional loss leads to p16INK4a promoter derepression and overexpression, shifting cells into replicative senescence, and preventing further cell cycling.26, 27 A large body of published data in the context of oropharyngeal and supraglottic squamous cell carcinoma confirm that p16INK4a overexpression predicts increased radiosensitivity and improved survival.11, 12, 13, 14, 15, 16, 17, 18 Published data specific to oral cavity squamous cell carcinoma examining p16INK4a, HPV status, and outcome are more limited.
Most HPV/p16INK4a studies include oral cavity squamous cell carcinoma among other head and neck anatomic sites and are underpowered to observe any association when limited to oral cavity alone.28 There is a wide range in the reported frequencies of strong and diffuse p16INK4a overexpression in oral cavity squamous cell carcinoma.29, 30, 31, 32, 33, 34, 35, 36, 37, 38 Here, in studying a large cohort of patients with oral cavity squamous cell carcinoma, we confirm that p16INK4a overexpression is uncommon (15.5%). Furthermore, we demonstrate that p16INK4a overexpression has a protective effect with respect to disease progression (hazard ratio 0.11, 95% CI 0.02–0.63). Our data are consistent with those of other large studies; Lingen et al30 and Jayasurya et al29 demonstrated p16INK4a overexpression in 11.2% of 409 patients, and 15.8% of 448 patients, respectively. A number of oral cavity squamous cell carcinoma studies confirm a direct relationship between p16INK4a and outcome.29, 32, 33, 34, 39
We also demonstrate that p16INK4a status does not correlate with transcriptionally active HPV16 for oral cavity squamous cell carcinoma; 20 of 24 (83%) HPV16-positive oral cavity squamous cell carcinoma did not overexpress p16INK4a. This is likely because of promoter methylation or homozygous deletion of CDKN2A; somatic point mutations of CDKN2A are very rare.37, 38, 39, 40, 41, 42 Conversely, 11 of 80 (13.8%) oral cavity squamous cell carcinoma overexpressed p16INK4a, yet harbored neither HPV16 nor HPV18 (HPV18 data not shown). The mechanism promoting p16INK4a overexpression in these circumstances is unknown. Importantly, classification of HPV status based on DNA assays and p16INK4a IHC expression can be misleading. HPV DNA+/ p16INK4a IHC-negative tumors are usually classified as harboring ‘passenger HPV’. We demonstrate that this profile does not exclude the possibility of transcriptionally active HR-HPV in the presence of silenced p16INK4a.
The weighted prevalence of HPV DNA in 4195 oral cavity squamous cell carcinoma patients is 20.2%, (95% CI 16.0–25.2), most commonly consisting of HPV16.28 There is a paucity of published data on HR-HPV E6/E7 RNA expression in oral cavity squamous cell carcinoma, directly relevant to virally mediated carcinogenesis.22, 23, 30, 43, 44, 45 Here we show that HPV16 and HPV18 are present in 22.6% (35/155) and 11% (13/119) of patients, respectively, consistent with the weighted prevalence of HPV DNA. Recently, a large study demonstrated a lower prevalence of HR-HPV E6/E7 expression that was demonstrated in 24/409 cases (5.9%, 95% CI 3.6–8.2).30
Three published studies report that HPV+ oral cavity squamous cell carcinoma are associated with poorer prognosis; two studies are from the same group in Taiwan and include an expanded cohort.46, 47, 48 The strengths of the combined Taiwanese study are the large patient numbers and multivariate analysis; study weaknesses include the lack of p16INK4a analysis and absence of validation for the HPV genotype data. The Taiwan study demonstrated an overall HPV prevalence of 21.2% among 410 patients; HPV16 was the most common subtype followed by HPV18.47 This group reported that HR-HPV was associated with decreased disease-free and disease-specific survivals at 2 years. Racial differences may contribute to these unusual findings. Curiously, they also found a significant association between low-risk HPV (HPV42, HPV43, HPV44, HPV53, HPV62, HPV66, HPV70, HPV71, HPV72, and HPV81) and distant metastases. As mentioned, HPV was genotyped using only a blot hybridization assay, and not validated.47 Duray et al48 reported on 147 patients with oral cavity squamous cell carcinoma; HPV was detected by PCR with consensus and type-specific primers. HR-HPV was associated with decreased disease-free survival on Kaplan–Meier analysis; no multivariate analysis was performed.48 Two studies showed nonsignificant trends of improved survival;49, 50 and three other studies found no association between HPV and outcome.35, 51, 52
Enhanced local adaptive immunity is known to have a protective impact on outcome in oral cavity squamous cell carcinoma and directly associates with cytotoxic CD8 cells.3, 4, 53 Here we show that HPV16/18 in oral cavity squamous cell carcinoma is not associated with enhanced local adaptive immunity. In contrast, we and others have demonstrated enhanced local immune response in HPV-mediated oropharyngeal squamous cell carcinoma.54, 55 Circulating and intratumoral HPV16-specific T cells are significantly increased in patients with HPV-mediated oropharyngeal squamous cell carcinoma.56, 57 Taken together, our data support that transcriptionally active HPV16 in oral cavity squamous cell carcinoma does not affect outcome, as two protective mechanisms for HPV-mediated oropharyngeal squamous cell carcinoma (p16INK4a overexpression and enhanced tumor immunity) are not relevant in this context.
It was the histological differences in patterns of invasion (POI) seen in HPV-mediated oropharyngeal squamous cell carcinomas versus oral cavity squamous cell carcinoma that led us to test the hypothesis that HPV oncoproteins, E6/E7, and downstream p16INK4a affect tumor invasion. The role of p16INK4a as an inhibitor of tumor invasion is not as well appreciated as its other tumor suppressor functions. Decreased glioma invasion was demonstrated with p16INK4a transfection.58 This change was not merely because of p16INK4a-mediated growth arrest, as transfection with p21Waf1, another cyclin-dependent kinase inhibitor, also induced G1 arrest but did not affect invasion.58 Similarly, overexpressing p16INK4a in A549 lung cancer cells suppressed MMP-2, yet transfection with p27Kip1, another cyclin-dependent kinase inhibitor, did not affect MMP-2.59 Overexpressing p16INK4a in breast cancer cells similarly decreases invasion under hypoxic conditions via HIF pathway suppression.60, 61, 62 Moreover, migration and invasion of cancer-associated stromal fibroblasts is enhanced by p16INK4a silencing, through protein kinase Akt and VEGF-A.63 Taken together, these data support the concept that p16INK4a can specifically regulate invasion pathways.
By manipulating p16INK4a expression in cancer cell lines derived from oral cavity squamous cell carcinoma with WPOI-5, which represents the most aggressive pattern of invasion,2, 3, 4 we show that p16INK4a overexpression decreases tumor invasiveness by modulating a number of ECM remodeling genes. Interestingly, different gene expression and kinomic profile response patterns were observed for the two cell lines, even though the magnitude of dampened invasion was similar for both; this speaks to the expected heterogeneity of the aggressive invasive phenotype and expression profile. For instance, p16INK4a overexpression silenced MMP1 in UAB-4, but upregulated MMP1 in UAB-3. Similarly, MMP3 had reciprocal response patterns to p16INK4a overexpression in the two cell lines.
We looked at genes with response patterns to p16INK4a overexpression and silencing with fold change of ≤0.66 or ≥1.5 and reciprocal (gene silencing and overexpression, or overexpression and silencing, in response to p16INK4a overexpression and silencing, respectively). Seven potentially interesting genes were common to both cell lines; three are directly involved in extracellular matrix remodeling: TIMP3, COL1A1, and PDGFRA. Three other common genes (FOLR1, FLK-1, and LYN) have also been linked to invasion.64, 65, 66 FOLR1 encodes FRα, folate receptor-α; stable FRα knockdown decreases invasiveness in ovarian carcinoma cell lines.64 FLK-1 (also known as KDR, VEGFR, VEGFR2) encodes a VEGF receptor tyrosine kinase; VEGF neutralization or knockdown decreases invasiveness of lung adenocarcinoma cell lines.65 LYN encodes Lyn (Lck/Yes novel tyrosine kinase), a member of the Src family of protein tyrosine kinases (SFK); Lyn silencing decreases migration and invasion of head and neck squamous cell carcinoma cell lines.66 Recently, Basu et al67 established tumor cell lines from patient-derived xenografts originally procured from oral squamous carcinomas with ‘low-risk and high-risk invasion patterns’ and showed that patient-derived xenografts from high-risk cancers contained more evidence of epithelial–mesenchymal transformation. These epithelial–mesenchymal-transformed cells were resistant to EGFR-targeted therapy. Thus, it would be interesting to compare patient response with EGFR inhibition with tumor pattern of invasion.
It is interesting that gene expression was predominantly reduced by p16INK4a overexpression, whereas kinase activity was predominantly increased. On closer inspection of individual cell lines, we noted that the UAB-3-p16INK4a cells had a preponderance of receptor tyrosine kinase (RTK) activity, whereas the UAB-4-p16INK4a was more related to nonreceptor tyrosine kinases (NRTKs). Although both RTKs and NRTKs have been associated with head and neck cancers, NRTKs are often linked with integrin signaling, well known for affecting migration/invasion.68, 69 Our integrated network modeling approach confirmed distinct signaling networks at work in these cell lines despite the very similar phenotype. Further evaluation of the signaling cascades at work within these model systems will help elucidate the mechanism by which p16INK4a promotes better outcomes and may also reveal novel targets for therapeutic development.
In summary, we present clinical data from a large cohort of oral cavity squamous cell carcinoma patients with respect to transcriptionally active HPV16, p16INK4a overexpression, local adaptive tumor immunity, and outcome. We demonstrate that HPV16 in oral cavity squamous cell carcinoma correlates with neither p16INK4a overexpression nor enhanced local tumor immunity, in contrast to HPV+ oropharyngeal squamous cell carcinomas that typically overexpress p16INK4a and demonstrate enhanced acquired tumor immunity. Multivariate modeling in our oral cavity squamous cell carcinoma cohort demonstrates a protective effect with p16INK4a overexpression and disease progression. Histological differences in patterns of invasion seen in HPV-mediated oropharyngeal squamous cell carcinomas versus oral cavity squamous cell carcinoma led us to test the hypothesis that HPV E6/E7, and downstream p16INK4a, affect tumor invasion. First, we demonstrated that primary carcinoma cell lines derived from oropharyngeal squamous cell carcinomas are less invasive than SCC cell lines from oral cavity squamous cell carcinoma. Overexpressing p16INK4a caused reproducible, dramatically decreased invasiveness in UAB-3 and UAB-4, two WPOI-5 oral cavity squamous carcinoma cell lines. Integrating p16INK4a-responsive RNA expression and kinase profiling data yielded a number of interesting p16INK4a-responsive invasion-related genes, whereas kinomic profiling revealed signaling networks that may be driving these invasion-related processes. Taken together, the data support that p16INK4a overexpression is protective for patients with oral cavity squamous cell carcinoma, unrelated to transcriptionally active HPV, and support the role of p16Ink4A protein as an inhibitor of invasion for head and neck squamous cell carcinoma.
References
Jemal A, Siegel R, Ward E et al. Cancer statistics. CA Can J Clin 2009;59:225.
Brandwein-Gensler M, Teixeira M, Lewis C et al. Oral squamous cell carcinoma: histological risk assessment, but not margin status, is strongly predictive of local disease-free and overall survival. Am J Surg Pathol 2005;29:167–178.
Brandwein-Gensler M, Smith RV, Wang B et al. Validation of the histological risk model in a new patient cohort with primary head and neck squamous cell carcinoma. Am J Surg Pathol 2010;34:676–688.
Li Y, Bai S, Carroll W et al. Validation of the risk model: high-risk classification and tumor pattern of invasion predict outcome for patients with low-stage oral squamous cell carcinoma. Head Neck Pathol 2013;7:211–223.
Panwar A, Batra R, Lydiatt WM et al. Human papillomavirus positive oropharyngeal squamous cell carcinoma: a growing epidemic. Cancer Treat Rev 2014;40:215–219.
Smith EM, Ritchie JM, Summersgill KF et al. Age, sexual behavior and human papillomavirus infection in oral cavity and oropharyngeal cancers. Int J Cancer 2004;108:766–772.
Gillison ML, Koch WM, Capone RB et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 2000;92:709–720.
Ang KK, Harris J, Wheeler R et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med 2010;363:24–35.
Fakhry C, Westra WH, Li S et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst 2008;100:261–269.
Weinberger PM, Yu Z, Haffty BG et al. Molecular classification identifies a subset of human papillomavirus–associated oropharyngeal cancers with favorable prognosis. J Clin Oncol 2006;24:736–747.
Lassen P, Eriksen JG, Hamilton-Dutoit S et al. Effect of HPV-associated p16INK4A expression on response to radiotherapy and survival in squamous cell carcinoma of the head and neck. J Clin Oncol 2009;27:1992–1998.
Lassen P, Eriksen JG, Hamilton-Dutoit S et al. HPV-associated p16-expression and response to hypoxic modification of radiotherapy in head and neck cancer. Radiother Oncol 2010;94:30–35.
Schwarz JK, Lewis JS Jr, Pfeifer J et al. Prognostic significance of p16 expression in advanced cervical cancer treated with definitive radiotherapy. Int J Radiat Oncol Biol Phys 2012;84:153–157.
Lau HY, Brar S, Klimowicz AC et al. Prognostic significance of p16 in locally advanced squamous cell carcinoma of the head and neck treated with concurrent cisplatin and radiotherapy. Head Neck 2011;33:251–256.
Rischin D, Young RJ, Fisher R et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J Clin Oncol 2010;28:4142–4148.
Kumar B, Cordell KG, Lee JS et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J Clin Oncol 2008;26:3128–3137.
Lewis JS Jr, Thorstad WL, Chernock RD et al. p16 positive oropharyngeal squamous cell carcinoma: an entity with a favorable prognosis regardless of tumor HPV status. Am J Surg Pathol 2010;34:1088–1096.
Witkiewicz AK, Knudsen KE, Dicker AP et al. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 2011;10:2497–2503.
Edge SB, Byrd DR, Comptom CC, Fritz AG, Greene FL, Trotti A, (eds) AJCC Cancer Staging Handbook 7th edn Springer: New York, 2010, pp 49, 50, 63 64.
Isayeva T, Xu J, Dai Q et al. African Americans with oropharyngeal carcinoma have significantly poorer outcomes despite similar rates of human papillomavirus-mediated carcinogenesis. Hum Pathol 2014;45:310–319.
Isayeva T, Said-Al-Naief N, Ren Z et al. Salivary mucoepidermoid carcinoma: demonstration of transcriptionally active human papillomavirus 16/18. Head Neck Pathol 2013;7:135–148.
Schlecht NF, Brandwein-Gensler M, Nuovo GJ et al. A comparison of clinically utilized human papillomavirus detection methods in head and neck cancer. Mod Pathol 2011;24:1295–1305.
Smeets SJ, Braakhuis BJ, Abbas S et al. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene 2006;25:2558–2564.
Braakhuis BJ, Snijders PJ, Keune WJ et al. Genetic patterns in head and neck cancers that contain or lack transcriptionally active human papillomavirus. J Natl Cancer Inst 2004;96:998–1006.
Ragin CC, Taioli E, Weissfeld JL et al. 11q13 amplification status and human papillomavirus in relation to p16 expression defines two distinct etiologies of head and neck tumours. Br J Cancer 2006;95:1432–1438.
Li H, Wang W, Liu X et al. Transcriptional factor HBP1 targets P16(INK4A), upregulating its expression and consequently is involved in Ras-induced premature senescence. Oncogene 2010;29:5083–5094.
Gil J, Peters G . Regulation of the INK4b–ARF–INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006;7:667–677.
Isayeva T, Li Y, Maswahu D et al. Human papillomavirus in non-oropharyngeal head and neck cancers: a systematic literature review. Head Neck Pathol 2012;6:S104–S120.
Jayasurya R, Sathyan KM, Lakshminarayanan K et al. Phenotypic alterations in Rb pathway have more prognostic influence than p53 pathway proteins in oral carcinoma. Mod Pathol 2005;18:1056–1066.
Lingen MW, Xiao W, Schmitt A et al. Low etiologic fraction for high-risk human papillomavirus in oral cavity squamous cell carcinomas. Oral Oncol 2013;49:1–8.
Jayasurya R, Francis G, Kannan S et al. p53, p16 and cyclin D1: molecular determinants of radiotherapy treatment response in oral carcinoma. Int J Cancer 2004;109:710–716.
Bova RJ, Quinn DI, Nankervis JS et al. Cyclin D1 and p16INK4A expression predict reduced survival in carcinoma of the anterior tongue. Clin Cancer Res 1999;5:2810–2819.
Chen YW, Kao SY, Yang MH . Analysis of p16INK4a Expression of oral squamous cell carcinoma in Taiwan: prognostic correlation without relevance to betel quid consumption. J Surg Oncol 2012;106:149–154.
Harris SL, Thorne LB, Seaman WT et al. Association of p16(INK4a) overexpression with improved outcomes in young patients with squamous cell cancers of the oral tongue. Head Neck 2011;33:1622–1627.
Reuschenbach M, Kansy K, Garbe K et al. Lack of evidence of human papillomavirus-induced squamous cell carcinomas of the oral cavity in southern Germany. Oral Oncol 2013;49:937–942.
Duncan LD, Winkler M, Carlson ER et al. p16 immunohistochemistry can be used to detect human papillomavirus in oral cavity squamous cell carcinoma. J Oral Maxillofac Surg 2013;71:1367–1375.
Sailasree R, Abhilash A, Sathyan KM et al. Differential roles of p16INK4a and p14ARF genes in prognosis of oral carcinoma. Cancer Epidem Biomarkers Prev 2008;17:414–420.
Dong Y, Wang J, Dong F et al. The correlations between alteration of p16 gene and clinicopathological factors and prognosis in squamous cell carcinomas of the buccal mucosa. J Oral Pathol Med 2012;41:463–469.
Su PF, Huang WL, Wu HT et al. p16(INK4A) promoter hypermethylation is associated with invasiveness and prognosis of oral squamous cell carcinoma in an age-dependent manner. Oral Oncol 2010;46:734–739.
O'Regan EM, Toner ME, Smyth PC et al. Distinct array comparative genomic hybridization profiles in oral squamous cell carcinoma occurring in young patients. Head Neck 2006;28:330–338.
O'Regan EM, Toner ME, Finn SP et al. p16(INK4A) genetic and epigenetic profiles differ in relation to age and site in head and neck squamous cell carcinomas. Hum Pathol 2008;39:452–458.
Yakushiji T, Noma H, Shibahara T et al. Analysis of a role for p16/CDKN2 expression and methylation patterns in human oral squamous cell carcinoma. Bull Tokyo Dent Coll 2001;42:159–168.
Boscolo-Rizzo P, Da Mosto MC, Fuson R et al. HPV-16 E6L83 V variant in squamous cell carcinomas of the upper aerodigestive tract. J Cancer Res Clin Oncol 2009;135:559–566.
Badaracco G, Rizzo C, Mafera B et al. Molecular analyses and prognostic relevance of HPV in head and neck tumours. Oncol Rep 2007;17:931–939.
Smeets SJ, Hesselink AT, Speel EJ et al. A novel algorithm for reliable detection of human papillomavirus in paraffin embedded head and neck cancer specimen. Int J Cancer 2007;121:2465–2472.
Lee LA, Huang CG, Liao CT et al. Human papillomavirus-16 infection in advanced oral cavity cancer patients is related to an increased risk of distant metastases and poor survival. PLoS One 2012;7:e40767.
Lee LA, Huang CG, Tsao KC et al. Increasing rates of low-risk human papillomavirus infections in patients with oral cavity squamous cell carcinoma: association with clinical outcomes. J Clin Virol 2013;57:331–337.
Duray A, Descamps G, Decaestecker C et al. Human papillomavirus DNA strongly correlates with a poorer prognosis in oral cavity carcinoma. Laryngoscope 2012;122:1558–1565.
Kaminagakura E, Villa LL, Andreoli MA et al. High-risk human papillomavirus in oral squamous cell carcinoma in young patients. Int J Cancer 2012;130:1726–1732.
Sugiyama M, Bhawal UK, Kawamura M et al. Human papillomavirus-16 in oral squamous cell carcinoma: clinical correlates and 5-year survival. Br J Oral Maxillofac Surg 2007;45:116–122.
Smith EM, Rubenstein LM, Haugen TH et al. Complex etiology underlies risk and survival in head and neck cancer human papillomavirus, tobacco, and alcohol: a case for multifactor disease. J Oncol 2012;2012:571862.
Elango KJ, Suresh A, Erode EM et al. The role of human papillomavirus in oral tongue squamous carcinoma. Asian Pac J Cancer Prev 2011;12:889–896.
Maleki S, Schlecht NF, Keller C et al. Lymphocytic host response to oral squamous cell carcinoma: an adaptive T-cell response at the tumor interface. Head Neck Pathol 2011;5:117–122.
Jung AC, Guihard S, Krugell S et al. CD8-alpha T-cell infiltration in human papillomavirus-related oropharyngeal carcinoma correlates with improved patient prognosis. Int J Cancer 2013;32:E26–E36.
Wansom D, Light E, Thomas D et al. UM head neck SPORE Program. Infiltrating lymphocytes and human papillomavirus-16-associated oropharyngeal cancer. Laryngoscope 2012;122:121–127.
Heusinkveld M, Goedemans R, Briet RJ et al. Systemic and local human papillomavirus 16-specific T-cell immunity in patients with head and neck cancer. Int J Cancer 2012;131:E74–E85.
Hoffmann TK, Arsov C, Schirlau K et al. T cells specific for HPV16 E7 epitopes in patients with squamous cell carcinoma of the oropharynx. Int J Cancer 2006;118:1984–1991.
Chintala SK, Fueyo J, Gomez-Manzano C et al. Adenovirus-mediated p16/CDKN2 gene transfer suppresses glioma invasion in vitro. Oncogene 1997;15:2049–2057.
Wang CH, Chang HC, Hung WC . p16 inhibits matrix metalloproteinase-2 expression via suppression of Sp1-mediated gene transcription. J Cell Physiol 2006;208:246–252.
Li L, Lu Y . Inhibition of hypoxia-induced cell motility by p16 in MDA-MB-231 breast cancer cells. J Cancer 2010;1:126–135.
Shyu KG, Hsu FL, Wang MJ et al. Hypoxia-inducible factor 1alpha regulates lung adenocarcinoma cell invasion. Exp Cell Res 2007;313:118–1191.
Gilkes DM, Bajpai S, Chaturvedi P et al. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem 2013;288:10819–10829.
Al-Ansari MM, Hendrayani SF, Tulbah A et al. p16INK4a represses breast stromal fibroblasts migration/invasion and their VEGF-A-dependent promotion of angiogenesis through Akt inhibition. Neoplasia 2012;14:1269–1277.
Siu MK, Kong DS, Chan HY et al. Paradoxical impact of two folate receptors, FRα and RFC, in ovarian cancer: effect on cell proliferation, invasion and clinical outcome. PLoS One 2012;7:e47201.
Roybal JD, Zang Y, Ahn YH et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res 2011;9:25–35.
Wheeler SE, Morariu EM, Bednash JS et al. Lyn kinase mediates cell motility and tumor growth in EGFRvIII-expressing head and neck cancer. Clin Cancer Res 2012;18:2850–2860.
Basu D, Bewley AF, Sperry SM et al. EGFR inhibition promotes an aggressive invasion pattern mediated by mesenchymal-like tumor cells within squamous cell carcinomas. Mol Cancer Ther 2013;12:2176–2186.
Kim SA, Kwon SM, Kim JA et al. 5'-Nitro-indirubinoxime, an indirubin derivative, suppresses metastatic ability of human head and neck cancer cells through the inhibition of Integrin β1/FAK/Akt signaling. Cancer Lett 2011;306:197–204.
Koontongkaew S, Amornphimoltham P, Monthanpisut P et al. Fibroblasts and extracellular matrix differently modulate MMP activation by primary and metastatic head and neck cancer cells. Med Oncol 2012;29:690–703.
Acknowledgements
This project was funded by the Department of Pathology, and an Intramural Pilot Program Award, Department of Radiation Oncology, University of Alabama at Birmingham.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Isayeva, T., Xu, J., Ragin, C. et al. The protective effect of p16INK4a in oral cavity carcinomas: p16Ink4A dampens tumor invasion—integrated analysis of expression and kinomics pathways. Mod Pathol 28, 631–653 (2015). https://doi.org/10.1038/modpathol.2014.149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.149
This article is cited by
-
Association between human papillomavirus and oral cancer: a literature review
International Journal of Clinical Oncology (2023)
-
The expression and prognostic impact of proinflammatory cytokines and their associations with carcinogens in oropharyngeal squamous cell carcinoma
Cancer Immunology, Immunotherapy (2020)
-
Expression and Significance of Cytokeratin 7, a Squamocolumnar Junction Marker, in Head and Neck Squamous Cell Carcinoma
Head and Neck Pathology (2018)
-
Cancer stage and pack-years, but not p16 or HPV, are relevant for survival in hypopharyngeal and laryngeal squamous cell carcinomas
European Archives of Oto-Rhino-Laryngology (2018)
