
Abstract
The switch/sucrose non-fermentable (SWI/SNF) subunit ARID1A (AT-rich interactive domain 1A gene) has been recently postulated as a novel tumor suppressor of gynecologic cancer and one of the driver genes in endometrial carcinogenesis. However, specific relationships with established molecular alterations in endometrioid endometrial cancer (EEC) are currently unknown. We analyzed the expression of ARID1A in 146 endometrial cancers (130 EECs and 16 non-EECs) in relation to alterations in the PI3K-Akt pathway (PTEN expression/KRAS/PIK3CA mutations), TP53 status (TP53 immunohistochemistry) and microsatellite instability. To discriminate between microsatellite instability due to somatic MLH1 promoter hypermethylation or germline mutations in one of the mismatch repair genes (Lynch syndrome), we included a ‘Lynch syndrome set’. This set included 21 cases with confirmed germline mutations and 15 cases that were suspected to have a germline mutation. Loss of ARID1A expression was exclusively found in EECs in 31% (40/130) of the EEC cases. No loss of expression of the other subunits of the SWI/SNF complex, SMARCD3 and SMARCB1, was detected. Alterations in the PI3K-Akt pathway were more frequent when ARID1A expression was lost. Loss of ARID1A and mutant-like TP53 expression was nearly mutually exclusive (P=0.0004). In contrast to Lynch-associated tumors, a strong association between ARID1A loss and sporadic microsatellite instability was found. Only five cases (14%) of the ‘Lynch syndrome set’ as compared with 24 cases (75%, P<0.0001) of the sporadic microsatellite-unstable tumors showed loss of ARID1A. These observations suggest that ARID1A is a causative gene, instead of a target gene, of microsatellite instability by having a role in epigenetic silencing of the MLH1 gene in endometrial cancer.
Similar content being viewed by others
Main
Recent genome-wide sequencing studies have demonstrated that the AT-rich interactive domain 1A (ARID1A) gene is frequently mutated in a wide variety of cancer types1 including a subset of gynecological cancers.2 In ovarian cancer, near half of the clear-cell subtype and 30% of the endometrioid subtype showed ARID1A mutations,3 particularly those that are related to endometriosis.4, 5 In endometrial cancer, mutations in the ARID1A gene have been reported in approximately 30% of both low- and high-grade endometrioid endometrial cancers (EECs) but not in serous endometrial carcinomas.6, 7, 8
ARID1A encodes a large nuclear protein involved in chromatin remodeling and interacts with several other proteins, including the core protein SMARCD3, to form the so-called switch/sucrose non-fermentable (SWI/SNF) complex. Remodeling activity is facilitated by ATP hydrolysis of SMARCD3, whereas the non-catalytic subunits of this complex, such as ARID1A and SNF5 (SMARCB1), are responsible for modulating the target specificity and activity of the ATPase. Chromatin remodeling complexes, like SWI/SNF, are master regulators of transcription factor action and enable gene transcription and silencing by facilitating or inhibiting transcription factors to bind to promoters and enhancers. Those complexes are involved in various processes that require alteration of chromatin structure, including DNA repair, DNA synthesis, mitosis and genomic instability.9, 10, 11 Most mutations in ARID1A are insertions or deletions, resulting in a truncated protein that is prone to rapid degradation. Therefore, ARID1A gene mutations are highly correlated with loss of protein expression as assessed by immunohistochemistry, even in the absence of mutation in the other allele (eg, haploinsufficiency).4, 7, 12 Loss of expression of ARID1A by immunohistochemistry can thus be used as a surrogate marker for the underlying gene mutation.
Endometrial cancer has traditionally been subdivided clinically, pathologically and molecularly into two subgroups. The most frequent (80%) endometrioid type (type 1) is characterized by mostly low- and intermediate-grade adenocarcinomas, morphologically resembling normal endometrioid glands and arising in a background of hyperestrogenism. Activation of the Wnt signaling pathway, P13K-Akt pathway activation by either PTEN loss, PIK3CA and KRAS mutations or a combination of those alterations, microsatellite instability (MSI) and less frequently TP53 mutational inactivation have been identified as the main molecular alterations driving endometrioid endometrial carcinogenesis. In contrast, about 20% of the endometrial tumors are high-grade non-endometrioid cancers (type 2), mostly of the serous or clear-cell subtypes, and are characterized by high frequency of mutations in TP53, p16 overexpression, HER2 amplification and global LOH.13
There is limited data concerning the relationship and interactions of ARID1A and the above-mentioned genetic alterations in (endometrial) cancer. In a recent study, it was demonstrated that ARID1A mutations frequently co-occur with mutations in the PIK3CA gene and with P13K-Akt pathway activation as defined by increased AKT1 activation in endometrial cancer,1 but the mutational status of the PIK3CA gene was not assessed. In gastric cancer, an association has been described between MSI and mutations in ARID1A.12, 14, 15, 16 As the rate of somatic ARID1A mutations in MSI cases was 12- to 61-fold higher than the global background mutation rate, it was suggested that the ARID1A gene is particularly targeted by MSI.12 A possible association between ARID1A loss and MSI, as has been reported for gastric cancer, has not been studied in endometrial cancer. Several studies1, 2, 12 have shown an inverse relationship between TP53 and ARID1A mutations, suggesting that mutations in either of these two genes result in a similar downstream effect. Liang et al1 showed in cell line models that ARID1A and TP53 may cooperate in one complex (ARID1A/SMARCD3/TP53), explaining why ARID1A and TP53 mutations may be mutually exclusive. However, a recent study of Rahman et al8 could not confirm this inverse relationship between ARID1A loss and TP53 mutational expression in a series of 111 endometrial cancers.
The aim of this study was therefore to determine the relationship between the expression of different SWI/SNF subunits (ARID1A, SMARCD3 and SMARCB1) to other previously identified molecular alterations in endometrial cancer to elucidate the role of inactivation of the SWI/SNF complex in endometrial cancer. For this purpose, we analyzed the expression of ARID1A, SMARCD3 and SMARCB1 in 146 endometrial cancers (130 endometrioid, 16 non-endometrioid) and related this to the PI3K-Akt pathway (PTEN expression and KRAS and PIK3CA mutation status) and TP53 expression. In addition, we studied the expression of ARID1A in sporadic microsatellite-unstable tumors and compared these findings with a set of microsatellite-unstable tumors (N=36) with a proven or suspected germline mismatch repair (MMR) defect (‘Lynch syndrome set’).
Materials and methods
Patients and Tissue Material
This analysis was performed on formalin-fixed, paraffin-embedded tumor tissue from 146 primary endometrial cancers (EECs, N=130; NEEC, N=16), referred to as the ‘primary study set’. This set was composed of cases collected at the Department of Pathology of the University Medical Center in Utrecht (N=70) and a selection of cases stored at the Leiden University Medical Center (N=76). Both sets had previously been used for publication.17, 18 Table 1 gives an overview of the clinicopathological features of this primary study set.
‘Lynch Syndrome Set’
To compare the sporadic microsatellite cases with endometrial cancers from Lynch syndrome patients, we included a set of tumors with either confirmed (n=21) or suspected (n=15) defects in the MMR genes, which have been collected and stored at the Leiden University Medical Center and previously used for publication.19, 20 All tumors in this group were microsatellite unstable and showed loss of protein expression of one or two of the MMR proteins MLH1, PMS2, MSH2 or MSH6. When MLH1 was affected, MLH1 promoter hypermethylation was ruled out using MS-PCR (see below). In 21 cases, a germline mutation could be identified. In 11 cases, a germline mutation was suspected, but could not be confirmed, mostly because the family did not want further genetic testing. Previous work has indicated that 90–95% of cases suspected for Lynch syndrome (as defined in this report by loss of expression of one of the MMR proteins and no promoter methylation of MLH1) have an underlying germline mutation in one of the MMR genes.21 After the analysis of the primary study set (see above), four patients with microsatellite-unstable tumors were suspected for an MMR germline mutation and were therefore included into the suspected for Lynch group and excluded from the sporadic microsatellite-unstable group. Clinicopathological features and other details of this ‘Lynch syndrome set’, including the type of germline mutation, can be found in Supplementary Table 1.
Immunohistochemistry
Immunohistochemistry was performed as described previously.18 Briefly, tumor blocks were sectioned (whole section) at a thickness of 4μm, dried, dewaxed and rehydrated. Endogenous peroxidases were inactivated by 0.3% H2O2/methanol. Antigen retrieval was achieved by microwave oven treatment in 10 mmol/l citrate buffer, pH 6.0, for staining of TP53. For staining of SMARCD3, SMARCB1, MLH1 and ARID1A, antigen retrieval was performed in 10 mmol/l Tris-EDTA, pH 9.0. Sections were incubated overnight with monoclonal SMARCD3 (clone G7, 1:400; Santa Cruz; sc-17796), monoclonal anti-BAF47/SNF5/SMARCB1 (amino acids 257–359, 1:100) (BD Transduction Lab; 61211), monoclonal ARID1A/BAF250a (amino acids 600–1018, 1:800; Santa Cruz; sc-32761), monoclonal MLH1 (clone ES05, 1:100; DAKO), monoclonal TP53 (clone DO-7; Neomarkers; Ab-5) and PTEN (clone 6.H2.1; 1:200; DAKO). The sections were then incubated for 30 min with a secondary antibody (Poly-HRP-GAM/R/R; DPV0110HRP; Immunologic). Diaminobenzidine tetrahydrochloride was used as a chromogen for SMARCB1, MLH1 and TP53. For SMARCD3 and ARID1A, we used DAB+ (DAKO; K3468). The slides were counterstained with hematoxylin.
Scoring Staining
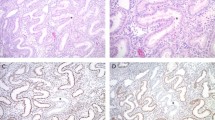
Blinded for patient characteristics and outcome of other analysis, slides were independently evaluated by two pathologists (TB and VTHBMS), and discrepancies were resolved by consensus under a double-headed microscope. TP53 was scored ‘wild-type-like’ when <50% of the tumor cells showed positive nuclear staining. TP53 was scored ‘mutant-like’ if >50% of the tumor cells showed strong positive nuclear staining, or when discrete geographical patterns showed >50% tumor cell positivity.17, 18 PTEN was scored negative when there was complete loss of expression in the tumor and positive stromal cells. PTEN staining was considered positive when strong, weak or heterogeneous staining was observed. MLH1 nuclear staining was scored as either positive or negative, with stromal and/or lymphocytic cells as internal controls. Nuclear staining intensity for SMARCD3, SMARCB1 and ARID1A was scored using a three-tier system as follows: loss of nuclear staining, weakly positive (low/moderate expression) and strongly positive (strong and diffuse expression). In two cases, ARID1A showed evenly distributed heterogeneous expression or a ‘checkerboard’ staining pattern of ARID1A. As previous studies have shown that mutations in the ARID1A gene with this expression pattern are absent,6, 8 we scored this pattern of expression as ‘moderate positive’. In 12 cases, focal, abrupt absence of nuclear staining was seen. This pattern has been referred to as a ‘clonal loss’ pattern and corresponds to mutations in ARID1A and therefore we scored these as ‘loss of expression’. Surrounding stromal cells served as positive internal controls (see Figure 1).

Examples of staining patterns observed for AT-rich interactive domain 1A (ARID1A) gene in endometrial cancer. Most endometrial cancer showed diffuse nuclear staining (a) and these were considered positive, in contrast to negative tumors that showed loss of ARID1A expression, with positive stromal cells as internal control (b). A subset of tumors showed a ‘checkerboard’ staining pattern and were considered positive (c). In addition, a ‘clonal loss’ pattern was observed (d), these cases were considered as loss of ARID1A expression.
Allele-Specific Hotspot Analysis
All samples were analyzed using a custom-made panel of hydrolysis probe assays, designed to detect three different PI3K hotspot mutations and seven different KRAS mutations.22 The hotspot mutations investigated for PI3K were exon 9, c.1624G>A; p.E542K and c.1633G.A; p.E545K and in exon 20 the c.3140A>G; p.H1047R. For KRAS exon 1, c.34G>A; p.G12S, c.34G>C; p.G12R, c.34G>T; p.G12C, c.35G>A; p.G12D, c.35G>C; p.G12A, c.35G>T; p.G12V and c.38G>A; p.G13D. Real-time qPCR was performed by allelic discrimination using primers and probes designed and ordered by Applied Biosystems.
Methylation-Specific PCR for MLH1
Methylation status of the MLH1 promoter was assessed using EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA) as described previously.23 In short, bisulfite modification of DNA (100 ng per sample) was carried out converting unmethylated cytosines, by a process of deamination, to uracil leaving the methylated cytosines unchanged. The PCR mix (25 μl volume) included: 50 ng bisulfite-converted DNA; 12 pM of forward and reverse primer; 10 × PCR buffer; 1.8 mM additional MgCl2, 200 μ M of each dNTP, 2.5 U Amplitaq Gold polymerase and nuclease-free water to make up to final volume. Cycling conditions were as follows: 90 °C for 5 min; 33 cycles of 92 °C for 45 s; 59 °C for 65 s; 72 °C for 45 s; 72 °C for 5 min; 4 °C hold. Primer sequences for the MLH1 promoter are available on request. As a readout system, we used gel electrophoresis.
Microsatellite Instability
From 76 tumors of the primary study set, the microsatellite status was previously analyzed,18 and from the remaining cases, MSI was assessed if either MLH1 or ARID1A expression was lost using the Promega MSI analysis system as described previously.18, 24 Tumors were classified as MSI-low as one marker showed an unstable pattern, or as MSI-high when more than one marker showed an unstable pattern. If all markers showed a normal pattern, the tumor was classified as microsatellite stable (MSS). In ambiguous cases, the experiment was repeated in combination with DNA extracted from unaffected myometrium to exclude biallelicity of the marker in normal tissue. Analysis of MSI was performed using PCR and subsequent fragment analysis on an automated capillary sequence machine, ABI 3130. These criteria for MSI are based on studies of colorectal carcinomas and have been used by us and others on endometrial cancer.18
Statistics
Analysis of ARID1A alteration frequencies was performed using Fisher’s exact test and P<0.05 was considered statistically significant.
Results
Expression of the SWI/SNF Subunits ARID1A, SMARCB1 and SMARCD3 in Endometrial Cancer
In total, whole sections of 146 endometrial cancers were used to analyze the expression profile of ARID1A, SMARCB1 and SMARCD3 (Table 1 and Figure 2). We found loss of ARID1A expression in 27% (N=40) and all these cases displayed an endometrioid morphology, implicating ARID1A as a marker for the endometrioid subtype (Figures 2a and b). Among the positive ARID1A cases, 59% showed high expression and 14% showed low to moderate expression. Almost all cases showed strong nuclear SMARCB1 expression (N=143, 98%) and no difference in staining was found between non-endometrioid and endometrioid cases, indicating that SMARCB1 probably does not have an important role in endometrial cancer. SMARCD3 was highly expressed in 118 cases (80%), but a significant proportion of cases (20%, N=28) showed decreased expression as compared with strong expression of normal endometrial glands in the same section (Figure 2d). Complete loss of SMARCD3 expression was not observed. Interestingly, a reduced SMARCD3 expression was three times more frequent in ARID1A-negative cases (P=0.0016; Figure 2b).

Expression of switch/sucrose non-fermentable (SWI/SNF) subunits AT-rich interactive domain 1A (ARID1A), SMARCB1 and SMARCD3 in endometrial cancer as determined by immunohistochemistry (N=146). (a) Frequencies of loss, low and high expression of the subunits ARID1A, SMARCB1 and SMARCD3. (c) Low SMARCD3 expression is significantly correlated with loss of ARID1A expression (12 and 38%, P=0.0016). (b) Example of an endometrioid endometrial cancer with loss of expression of ARID1A (immunohistochemistry). (d) Same case with low SMARCD3 expression. The inset shows normal SMARCD3 expression of the non-neoplastic adjacent endometrial glands.
Relationship Between ARID1A Expression and PI3K-Akt Pathway Alterations
To determine the relationship between ARID1A expression and alterations in the PI3K-Akt axis, we investigated the expression of PTEN and analyzed the mutational status of KRAS and PIK3CA in all tumors of the primary study set (Table 2). We found 47% loss of PTEN expression, 20% KRAS mutations and 18% PIK3CA hotspot mutations in the primary study set. There was no significant correlation between loss of ARID1A and any of the independent molecular alterations of the PI3K-Akt axis; however, a trend was observed for more alterations in the group of tumors that showed loss of ARID1A expression. Furthermore, the number of cases that showed no alterations in any of the tested parameters of the PI3K-Akt pathway was 4.6-fold higher in tumors with normal ARID1A expression (P=0.042; Table 2).
Relationship Between ARID1A Expression and TP53 Status
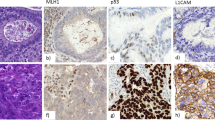
Examples of wild-type- and ‘mutant’-like TP53 staining in relation to the ARID1A expression in both endometrioid and non-endometrioid cancers are shown in Figure 3a. In a previous study,18 we have shown a high concordance rate between a ‘mutant’-like pattern and the presence of TP53 mutations. Of the 30 tumors with ‘mutant’-like TP53 staining, 10 were of non-endometrioid morphology and 20 showed endometrioid morphology. Only 1 of the 30 tumors with a ‘mutant’-like TP53 staining displayed loss of ARID1A. This tumor showed endometrioid morphology and the TP53 mutation was confirmed by Sanger sequencing (data used from previous paper18). All non-endometrioid cancers (independent of TP53 status) were ARID1A positive, including two clear-cell carcinomas. Figure 3b shows the frequency of TP53 mutant-like staining in the ARID1A-positive (27%) and the ARID1A-negative group (2.5%). These data clearly indicate an inverse relationship between ARID1A loss and mutations in TP53 in endometrial cancer, independent of tumor morphology.


Inverse relationship between TP53 status and AT-rich interactive domain 1A (ARID1A) expression in endometrial cancer. (a) Three examples (two endometrioid endometrial carcinoma and one serous carcinoma of the endometrium) exemplify this inverse relationship. (A1 and 2) An endometrioid endometrial carcinoma with loss of ARID1A expression and wild-type TP53 immunohistochemistry. (A3 and 4) An endometrioid endometrial carcinoma with positive ARID1A expression and TP53 mutant-like expression. (A5 and 6) A serous carcinoma with positive ARID1A expression and TP53 mutant-like staining. TP53 mutations were confirmed by Sanger sequencing. (b) Incidence of TP53 mutant staining pattern in relation to ARID1A status determined by immunohistochemistry. Solid/black area represents TP53 mutant-like staining, whereas open/white area represents TP53 wild-type staining pattern. Rates of TP53 mutant-like expression are indicated (27 and 2.5%, P<0.0004).
Relationship Between MSI and ARID1A Expression
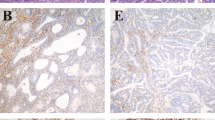
Furthermore, we investigated the relationship between MSI and the expression of ARID1A in the primary study set (Figure 4). We found MSI in 11 tumors with positive ARID1A expression (10%), as compared with 25 MSI tumors in the group that showed loss of ARID1A expression (63%, P<0.0001; Figure 4a). An example of the ARID1A staining in a MSS and unstable tumor is depicted in Figure 4b.

Relationship of AT-rich interactive domain 1A (ARID1A) expression and microsatellite instability (MSI). (a) Incidence of MSI in ARID1A-positive and -negative cases, as determined by immunohistochemistry. Solid/black area indicates microsatellite-unstable cases (MSI), whereas open/white area represents microsatellite-stable cases (MSS). Rates of MSI in the groups are indicated (10 and 63%, P<0.0001). (b) Two examples illustrating the inverse relationship of ARID1A expression in a MSS (B1) and an unstable (B2) case.
Next, a subset analysis was performed on MSI tumors. We determined whether the MSI was due to germline mutations or due to promoter hypermethylation of MLH1 by MS-PCR and expression analysis of the MMR genes. In 89% (N=32) of the MSI cases from the primary study set, loss of MLH1 expression was coexisting with MLH1 promoter hypermethylation and these cases were considered ‘sporadic microsatellite unstable’. In four cases, a germline mutation in one of the MMR genes was suspected and these were added to the ‘suspected for germline mutation’ group. We then compared the expression of ARID1A in the tumors with sporadic MSI to the microsatellite-unstable tumors because of proven or suspected germline mutations (Figure 5a). In this comparison, we found loss of ARID1A expression in only five cases (14%) of the ‘Lynch syndrome set’, as compared with 24 cases (75%, P<0.0001) of the sporadic microsatellite-unstable tumors. This difference could not be explained by TP53 status, as these groups only harbored one case with mutant TP53 expression.

Relationship of AT-rich interactive domain 1A (ARID1A) expression and microsatellite instability (MSI) subdivided in sporadic MSI and MSI because of germline mutations. (a) Loss of ARID1A expression was observed in 75% of the MSI tumors because of MLH1 (MutL homolog 1) promoter hypermethylation, whereas only 13 and 14% of the MSI tumors because of suspected or confirmed germline mutation (P<0.0001). (b) Case of endometrioid endometrial cancer with loss of ARID1A expression and MLH1 expression because of promoter hypermethylation of the MLH1 gene (#, two bands visible in the methylation-specific polymerase chain reaction (MSP-PCR), m (methylated), um (unmethylated), with a positive and negative control and H2O control). (c) Case of endometrioid endometrial cancer in a patient with Lynch syndrome with normal expression of ARID1A but loss of MLH1 expression because of a germline mutation in the MLH1 gene (c.806C>G,p.Ser269X).
Discussion
In this study, we analyzed the association of ARID1A loss with other components of the SWI/SNF complex, including SMARCD3 and SMARCB1, and related these findings to established molecular alterations, such as mutational inactivation of TP53, PI3K-Akt pathway activation and MSI in endometrial carcinogenesis. We found loss of ARID1A expression exclusively in endometrioid endometrial carcinomas. SMARCB1 expression was not affected in any of the endometrial cancer studied, whereas SMARCD3 expression was reduced in a subset of tumors, particularly those that showed loss of ARID1A. Furthermore, ARID1A loss frequently co-occurred with alterations in the PI3K-Akt pathway, and the frequency of alterations in this signaling pathway was much lower in those cases with normal ARID1A expression. Mutant TP53 expression showed an inverse relationship with loss of ARID1A expression, independent of tumor morphology. Finally, loss of ARID1A expression was significantly associated with sporadic MSI but not with MSI due to germline mutations.
From the investigated SWI/SNF subunits, only ARID1A was found to be completely negative in a subset of cases. The frequency of ARID1A loss in the primary study set (27%) corresponds to other recent publications6, 25 and argues in favor of a tumor suppressor role for ARID1A in endometrial cancer. We did not test the mutation status of ARID1A gene itself, but others have shown a strong correlation between the presence of ARID1A mutations and loss of ARID1A expression, as established by immunohistochemistry.4, 6, 12, 26 ARID1A loss was limited to tumors with endometrioid morphology and not found in non-endometrioid subtypes, consistent with previous studies.6 These data suggest that ARID1A loss is involved in the carcinogenesis of EECs and may be used as an additional marker in clinical diagnostics if morphological typing is difficult.27, 28
This study was the first to investigate the expression of two other relevant subunits of the SWI/SNF complex (SMARCB1 and SMARCD3) in relation to ARID1A. SMARCB1 and SMARCD3 inactivation has been observed in many different types of human cancer, including lung, pancreatic and kidney carcinomas and sarcomas.29, 30, 31, 32, 33 We found normal expression of SMARCB1 in all tumors and therefore conclude that this SWI/SNF subunit does not have an important role in endometrial carcinogenesis. Reduced SMARCD3 expression was significantly associated with ARID1A loss. This finding corresponds to a study that showed altered levels of SWI/SNF subunit expression in mouse ES cells with knockout of one or both alleles of ARID1A.34 Taken together, these data suggest that SMARCB1 and SMARCD3 expression are not independently affected in endometrial cancer, but the expression level may be reduced when the stability of the SWI/SNF complex is disturbed because of ARID1A loss.
In our analysis of the PI3K-Akt pathway, we found more alterations in this signal-transduction pathway when ARID1A was not expressed, as compared to tumors with normal ARID1A expression (Table 2). Similar observations were found by others in endometrial cancer,1 ovarian clear-cell carcinomas35 and gastric cancer.36 This may indicate cross-talk between ARID1A and the PI3K-Akt pathway. In vitro work using endometrial cancer cell lines supports this by showing increased AKT phosphorylation in response to ARID1A knockdown, demonstrating ARID1A as a regulator of the PI3K-Akt pathway in a similar manner as PTEN.1 On the basis of these findings, we speculate that co-occurrence of PIK3CA mutations and ARID1A loss may be explained by a mechanism of oncogenic addiction, in which mutational pressure on the PIK3CA gene is relatively high in ARID1A-mutated tumors. Understanding this relationship may become particularly important when targeting strategies are designed in the future.37
We found an inverse relationship between TP53 ‘mutant’-like expression and loss of ARID1A expression in endometrial cancer and found no loss of ARID1A in non-endometrioid endometrial carcinomas. In ovarian cancer, TP53 mutations were found to be mutually exclusive with ARID1A mutations,3 and in gastric cancer, a similar inverse relationship was identified.12 Rahman et al8 used a different TP53 scoring system (10% staining was considered positive) and did not find an association between ARID1A and TP53 expression in endometrial cancer. In our study, 30 tumors showed a ‘mutant’-like TP53 expression and all but 1 showed normal ARID1A expression. This finding is consistent with the hypothesis that ARID1A and TP53 work along the same pathway and the discrepancy with Rahman’s findings are likely the result of a difference in TP53 scoring. ARID1A may mediate its tumor suppressor function by interacting with TP53 to regulate CDKN1a and SMAD3 transcription to suppress tumor growth.2 Interestingly with respect to this hypothesis is that endometrial carcinomas with alterations in ARID1A or TP53 result in phenotypically different tumor types. Tumors with loss of ARID1A expression are endometrioid, whereas TP53-mutated tumors are predominantly of the non-endometrioid type.
We found a significant association between loss of ARID1A expression and MSI because of MLH1 promoter hypermethylation in endometrial cancer. In 75% of the sporadic microsatellite-unstable cases, ARID1A loss was observed in contrast to only 14% in cases with MSI because of germline mutations (Lynch syndrome). This association was not found in a recent immunohistochemical study including ovarian and endometrial cancers.8 The most likely explanation for this discrepancy is that immunohistochemistry alone does not discriminate between sporadic and germline MSI. Our findings support the hypothesis that MSI is preceded by ARID1A loss and argues against the existing hypothesis that ARID1A mutations are the result of MSI, as has been postulated for gastric cancer where a similar association between ARID1A mutations and MSI was found.12 In these studies, only the presence or absence of MSI was reported without the underlying cause (germline mutation or MLH1 promoter hypermethylation). We consider two hypotheses that may explain the association between ARID1A loss and sporadic MSI. Firstly, ARID1A loss may be the result of ARID1A promoter hypermethylation and not mutations, and the co-occurrence with MLH1 promoter hypermethylation is consequently a reflection of global genomic hypermethylation (CIMP). This explanation may be viable, as CpG islands have been identified on the ARID1A promoter and ARID1A promoter hypermethylation has been reported recently in invasive breast cancer.38 However, several other studies have shown that ARID1A loss of expression is strongly associated with mutations in the ARID1A gene, and for endometrial cancer, the mutation rate corresponds with the frequency of ARID1A loss.4, 6, 12, 26 Moreover, promoter hypermethylation as inactivating mechanism of the ARID1A gene has not been reported in endometrial cancer.39 An alternative hypothesis is that loss of ARID1A results in epigenetic alterations by a deficient SWI/SNF complex that may lead to MLH1 promoter hypermethylation. This could be achieved by a direct interaction of SWI/SNF on the MLH1 promoter as has been observed in rhabdoid tumors, where the loss of SNF5 (SMARCB1) resulted in promoter hypermethylation and loss of expression of the BIN-1 gene.40 These data support the idea that locus-specific epigenetic silencing is a commonality in tumors with dysfunctional SWI/SNF chromatin remodeling complex. Additional research focusing on the interaction between the SWI/SNF complex and the MLH1 promoter will be required to further delineate this mechanism and to better understand the intriguing association between loss of ARID1A expression and MLH1 promoter hypermethylation.
In conclusion, this study is the first to examine the relationship between loss of ARID1A expression and other important molecular alterations described in endometrioid endometrial carcinogenesis. We found that ARID1A loss is a relatively frequent (27%) event in EEC, and that the subunits of the SWI/SNF complex (SMARCD3 and SMARCB1) do not seem to have an important role. Loss of ARID1A and TP53 mutational inactivation was nearly mutually exclusive in endometrial cancer. Finally, our main finding is that loss of ARID1A expression is significantly associated with sporadic MSI but not with MSI because of germline mutation in one of the MMR genes. Although still speculative, this places ARID1A in a molecular pathway driving endometrioid endometrial carcinogenesis towards MSI through epigenetic alterations of the MLH1 gene. Further research will be required to delineate the exact mechanism by which ARID1A controls the epigenetic landscape in endometrial cancer.
References
Liang H, Cheung LW, Li J et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res 2012;22:2120–2129.
Guan B, Wang TL, I Shih . ARIDIA, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res 2011;71:6718–6727.
Xiao W, Awadallah A, Xin W . Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int J Clin Exp Pathol 2012;5:642–650.
Maeda D, Mao TL, Fukayama M et al. Clinicopathological significance of loss of ARID1A Immunoreactivity in ovarian clear cell carcinoma. Int J Mol Sci 2010;11:5120–5128.
Maeda D, Shih I . Pathogenesis and the role of ARID1A mutation in endometriosis-related ovarian neoplasms. Adv Anat Pathol 2013;20:45–52.
Guan B, Mao TL, Panuganti PK et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol 2011;35:625–632.
Wiegand KC, Lee AF, Al-Agha OM et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol 2011;224:328–333.
Rahman M, Nakayama K, Rahman MT et al. Clinicopathologic analysis of loss of AT-rich interactive domain 1A expression in endometrial cancer. Hum Pathol 2012;44:103–109.
Reisman D, Glaros S, Thompson EA . The SWI/SNF complex and cancer. Oncogene 2009;28:1653–1668.
Wilson BG, Roberts CW . SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011;11:481–492.
Wu JN, Roberts CW . ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 2013;3:35–43.
Wang K, Kan J, Yuen ST et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet 2011;43:1219–1223.
Zheng W, Xiang L, Fadare O et al. A proposed model for endometrial serous carcinogenesis. Am J Surg Pathol 2011;35:e1–e14.
Abe H, Maeda D, Hino R et al. ARID1A expression loss in gastric cancer: pathway-dependent roles with and without Epstein–Barr virus infection and microsatellite instability. Virchows Arch 2012;461:367–377.
Wang DD, Chen YB, Pan K et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS One 2012;7:e40364.
Jones S, Li M, Parsons DW et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat 2012;33:100–103.
Seeber LM, Zweemer RP, Marchionni L et al. Methylation profiles of endometrioid and serous endometrial cancers. Endocr Relat Cancer 2010;17:663–673.
Nout RA, Bosse T, Creutzberg CL et al. Improved risk assessment of endometrial cancer by combined analysis of MSI, PI3K-AKT, Wnt/beta-catenin and P53 pathway activation. Gynecol Oncol 2012;126:466–473.
de Leeuw WJ, Dierssen J, Vasen HF et al. Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J Pathol 2000;192:328–335.
Vasen HF, Hendriks Y, de Jong AE et al. Identification of HNPCC by molecular analysis of colorectal and endometrial tumors. Dis Markers 2004;20:207–213.
Vasen HF, Blanco I, Aktan-Collan K et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013;0:1–13.
van ER, Licht J, Schrumpf M et al. Rapid KRAS, EGFR, BRAF and PIK3CA mutation analysis of fine needle aspirates from non-small-cell lung cancer using allele-specific qPCR. PLoS One 2011;6:e17791.
van Roon EH, van PM, Middeldorp A et al. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC Cancer 2010;10:180.
Boland CR, Thibodeau SN, Hamilton SR et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248–5257.
Werner HM, Berg A, Wik E et al. ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Mod Pathol 2012;26:428–434.
Wiegand KC, Shah SP, Al-Agha OM et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010;363:1532–1543.
Bartosch C, Exposito MI, Lopes JM . Low-grade endometrial stromal sarcoma and undifferentiated endometrial sarcoma: a comparative analysis emphasizing the importance of distinguishing between these two groups. Int J Surg Pathol 2010;18:286–291.
Clarke BA, Gilks CB . Endometrial carcinoma: controversies in histopathological assessment of grade and tumour cell type. J Clin Pathol 2010;63:410–415.
Medina PP, Sanchez-Cespedes M . Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics 2008;3:64–68.
Reisman DN, Sciarrotta J, Wang W et al. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res 2003;63:560–566.
Wong AK, Shanahan F, Chen Y et al. BRG1, a component of the SWI–SNF complex, is mutated in multiple human tumor cell lines. Cancer Res 2000;60:6171–6177.
Dal MM, Hong SM, Hebbar S et al. Loss of expression of the SWI/SNF chromatin remodeling subunit BRG1/SMARCA4 is frequently observed in intraductal papillary mucinous neoplasms of the pancreas. Hum Pathol 2012;43:585–591.
Hollmann TJ, Hornick JL . INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 2011;35:e47–e63.
Gao X, Tate P, Hu P et al. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci USA 2008;105:6656–6661.
Yamamoto S, Tsuda H, Takano M et al. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol 2011;25:615–624.
Zhang X, Zhang Y, Yang Y et al. Frequent low expression of chromatin remodeling gene ARID1A in breast cancer and its clinical significance. Cancer Epidemiol 2012;36:288–293.
Korets SB, Czok S, Blank SV et al. Targeting the mTOR/4E-BP pathway in endometrial cancer. Clin Cancer Res 2011;17:7518–7528.
Zhang X, Sun Q, Shan M et al. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA rxpression in many invasive breast cancers. PLoS One 2013;8:e53931.
Tao MH, Freudenheim JL . DNA methylation in endometrial cancer. Epigenetics 2010;5:491–498.
McKenna ES, Tamayo P, Cho YJ et al. Epigenetic inactivation of the tumor suppressor BIN1 drives proliferation of SNF5-deficient tumors. Cell Cycle 2012;11:1956–1965.
Acknowledgements
This work was supported by the KWF Foundation of the Netherlands (UL2012-5447). We thank Michelle Osse (technician) for her assistance with the experiments and Carli Tops for providing the germline mutation annotations. Klaas van der Ham is acknowledged because of the microphotographs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article
Cite this article
Bosse, T., ter Haar, N., Seeber, L. et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol 26, 1525–1535 (2013). https://doi.org/10.1038/modpathol.2013.96
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.96
Keywords
This article is cited by
-
Multiomics analysis revealed the mechanisms related to the enhancement of proliferation, metastasis and EGFR-TKI resistance in EGFR-mutant LUAD with ARID1A deficiency
Cell Communication and Signaling (2023)
-
The effects of ARID1A mutation in gastric cancer and its significance for treatment
Cancer Cell International (2023)
-
Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response
Journal of Biomedical Science (2022)
-
ARID1A genomic alterations driving microsatellite instability through somatic MLH1 methylation with response to immunotherapy in metastatic lung adenocarcinoma: a case report
Journal of Medical Case Reports (2021)
-
BRG1 knockdown inhibits proliferation through multiple cellular pathways in prostate cancer
Clinical Epigenetics (2021)
