
Abstract
About 10–15% of gastrointestinal stromal tumors (GISTs) carry wild-type sequences in all hot spots of KIT and platelet-derived growth factor receptor alpha (PDGFRA) (wt-GISTs). These tumors are currently defined by having no mutations in exons 9, 11, 13, and 17 of the KIT gene and exons 12, 14, and 18 of the PDGFRA gene. Until now, the analysis of further exons is not recommended. However, we have previously published a report on a KIT exon 8 germline mutation, which was associated with familial GIST and mastocytosis. We therefore investigated whether KIT exon 8 mutations might also occur in sporadic GIST. We screened a cohort of 145 wt-GISTs from a total of 1351 cases from our registry for somatic mutations in KIT exon 8. Two primary GISTs with an identical exon 8 mutation (p.D419del) were detected, representing 1.4% of all the cases analyzed. Based on all GISTs from our registry, the overall frequency of KIT exon 8 mutations was 0.15%. The first tumor originating in the small bowel of a 53-year-old male patient had mostly a biphasic spindled-epithelioid pattern with a high proliferative activity (14 mitoses/50 HPF) combined with a second low proliferative spindle cell pattern (4/50 HPF). The patient developed multiple peritoneal metastases 29 months later. The second case represented a jejunal GIST in a 67-year old woman who is relapse-free under adjuvant imatinib treatment. We conclude that about 1–2% of GISTs being classified as ‘wild type’ so far might, in fact, carry KIT mutations in exon 8. Moreover, this mutational subtype was shown to be activating and imatinib sensitive in vitro. We therefore propose that screening for KIT exon 8 mutations should become a routine in the diagnostic work-up of GIST and that patients with an exon 8 mutation and a significant risk for tumor progression should be treated with imatinib.
Similar content being viewed by others


Main
Gastrointestinal stromal tumors (GISTs) are potentially malignant mesenchymal tumors of the gastrointestinal tract. KIT or — less often — PDGFRA (platelet-derived growth factor receptor alpha) mutations leading to a ligand-independent autoactivation of one of two tyrosine kinases are the oncogenetic drivers of these tumors.1, 2
In GIST, primary KIT mutations do not occur arbitrarily but only in the hot spot regions, which are exons 9, 11, 13, and 17. These encode an extracellular domain, the juxtamembrane domain, and the kinase domains 1 and 2, respectively. Corresponding affected exons of the closely related PDGFRA gene are 12, 14, and 18.3, 4, 5 Activating mutations in the remaining exons of both the genes have not been described so far in sporadic tumors. It is now widely accepted that GISTs are characterized by a close genotype–phenotype correlation in terms of location within the gastrointestinal tract, morphology, clinical behavior, and response to treatment with imatinib, which is a potent tyrosine kinase inhibitor. Thus, the mutational status has a high clinical impact as: (1) it can confirm the diagnosis of GIST in selected cases that are hard to diagnose; (2) it has a prognostic value; and (3) it is predictive for the response to molecular-targeted therapies, eg, with imatinib. Concerning the prognostic value, specific mutations in KIT exon 11 were shown to be associated with a higher risk of metastatic disease,6 whereas most PDGFRA-mutated GISTs show a less aggressive behavior.7, 8 In intestinal GISTs, KIT exon 9 mutations go along with aggressive behavior.9 In the context of targeted treatment, it has been recognized that the most common PDGFRA mutation (p.D842V), which nearly exclusively occurs in GISTs of the stomach, leads to resistance against imatinib.10 The detection of KIT exon 9 mutations in metastatic GISTs warrants doubling of the daily dose of imatinib to 800 mg/day in order to increase the response rate to treatment.11, 12 Thus, mutational testing of GIST is recommended in the diagnostic work-up of all GIST specimens, eg, by NCCN guidelines13 and the ESMO Clinical Practice Guidelines,14 at least in GISTs with intermediate or high risk of recurrence.
However, about 10–15% of GISTs carry wild-type sequences in all known hot spots of KIT and PDGFRA (wt-GISTs).15 In most cases, these wt-GISTs are known to have no or a poor response to tyrosine kinase inhibition. It now becomes apparent that wt-GISTs constitute an obviously heterogeneous group of different cancer subtypes. A subset of these tumors seem to be driven by other amplifications or oncogenic mutations of genes such as in IGF1R, BRAF, or PIK3CA.16, 17 Another subset of wt-GISTs arises in the context of hereditary syndromes. In the autosomal-dominant disorder, neurofibromatosis type 1 multiple wt-GISTs can occur,18, 19 arising predominantly in the small bowel. Patients with Carney–Stratakis syndrome suffer from gastric wt-GIST and paragangliomas.20 This syndrome is caused by germline mutations in the succinate dehydrogenase (SDH) subunits B, C, or D, leading to dysfunction of complex II of the electron transport chain.20, 21 However, it was shown that a subset of patients outside the Carney–Stratakis syndrome reveals mutations in SDH subtype B or C, pointing toward a pathogenetic role of SDH dysregulation in wt-GISTs.22 Paediatric GISTs, typically present in the stomach of young girls, display an epithelioid phenotype and can be multifocal. KIT and PDGFRA mutations are mostly absent23 and a subgroup of them is again attributable to succinate hydrogenase deficiency. Thus, the group of wt-GISTs is heterogeneous and further molecular characterization is needed to define subgroups, which might lead to a more specific therapy for these patients.
As stated earlier, wt-GISTs are currently defined by having wild-type sequences in exons 9, 11, 13, and 17 of the KIT gene and exons 12, 14, and 18 of the PDGFRA gene. Until now, the analysis of additional exons is not recommended in the routine molecular diagnostics of GISTs. However, we have previously published a report on a KIT exon 8 germline mutation,24 which was associated with systemic mastocytosis and familial GISTs.
The aim of this study was to investigate whether KIT exon 8 mutations also occur in sporadic GISTs. We therefore screened a cohort of 145 wt-GISTs for somatic mutations in KIT exon 8.
Materials and methods
Cases and Immunohistochemistry
A total of 145 wt-GISTs were extracted from our consultation files. All cases were wild type for KIT exons 9, 11, 13, and 17 and PDGFRA exons 12, 14, and 18. Baseline characteristics of the cases are displayed in Tables 1 and 2. The diagnosis of GISTs was made in all the presented cases by a senior pathologist specialized in soft tissue pathology (H-US, EW or RB). Immunohistochemistry for CD34 (clone QBEnd10, DAKO, 1:100), KIT (A4502, DAKO, 1:100), PDGFRA (SC-338, Santa Cruz, 1:50), DOG1 (Spring Bioscience, Fremont, CA, USA; 1:100) and Ki67 (Mib1, DAKO, 1:1000) was carried out as described earlier.25
Mutational Analysis
For genomic DNA-extraction, the lesional areas were marked on a haematoxylin and eosin-stained slide by a senior pathologist (H-US, EW, or RB). Subsequently, the tissue was scraped from unstained slides and DNA was extracted using standard methods as described previously.26 KIT exons 9, 11, 13, and 17 as well as PDGFRA exons 12, 14, and 18 DNA were amplified by PCR with specific primers and sequenced as described previously.26, 27 KIT exon 8 DNA was amplified by PCR with the following specific primers: Forward: 5′-GAAGTGAATGTTGCTGAGG-3′; and Reverse: 5′-GTGAATTGCAGTCCTTCC-3′. PCR and cycle sequencing were repeated twice.
Fluorescence In-Situ Hybridization (FISH) Analysis
FISH was performed on interphase nuclei of 4-μm sections of FFPE tissue. Locus-specific probe was derived from the BAC clone RP11-74L18 (imaGenes GmbH, Berlin, Germany) containing the full-length KIT gene using a Nick Translation Kit (Abbott Molecular, Germany) and spectrum orange-labelled dUTP (Abbott Molecular, Germany). KIT probe was used together with the chromosome 4 centromeric enumeration probe (CEP4, Abbott Molecular, Germany).
Deparaffinized slides were incubated in 0.2 M HCl for 20 min, followed by washing and 80 °C heat pretreatment in pretreatment solution (Abbott Molecular, Germany) for 30 min and further washing. Tissue was digested for 1.5 h with protease, slides were washed again, and fixed in 4% buffered formalin. All pretreatment steps were carried out in a prehybdridization processor (VP2000, Abbott Molecular). After washing, dehydration, and air drying, an appropriate amount of fluorochrome-labeled probe mixture was applied to the centre of the tissue section. Tissue DNA and probe mixture were co-denatured at 85 °C for 10 min, cooled down to 37 °C, and hybridized overnight in a Hybrite Hybridizer (Abbott Molecular, Germany). After stringent washing and dehydration, sections were counterstained using fluorescence mounting media containing de-amidino-phenyl-indole (DAPI, Zytovision GmbH, Germany). FISH slides were evaluated with a fluorescence microscope (DM 5500, Leica, Germany) using appropriate filter sets. Orange (KIT) and green (CEP4) signals were counted in 60 non-overlapping tumor cell nuclei from the three tumor areas.
Results
Clinicopathological Features
A total of 145 patients with proven wild-type sequences in all known mutational hot spots of KIT and PDGFRA were included in our study. Detailed clinicopathological data are provided in Table 1.
A comparison of these data with established clinicopathological features of GISTs (including wt-GISTs) demonstrates that our wt-GIST cases display a more or less representative cohort. For instance, small bowel GISTs (n=80) outnumbered GISTs of the stomach (n=53) and of the oesophagus and colon (n=1 each). Most cases displayed a spindled or mixed cell morphology (n=125/139) and only 14 cases revealed a pure epithelioid phenotype. In all, 12 patients were known to fulfill clinical criteria of neurofibromatosis type 1 and one patient presented with a Carney dyade. The tumor size was known in 121 cases and ranged from 0.1 to 35.0 cm in diameter (mean: 5.4 cm, median: 4.5 cm). Mitotic count was assessable in 136 cases and ranged from 0 to 90 (mean: 4.8; median: 1), with most cases having a low mitotic count. Therefore, according to the NCCN-AFIP criteria,28 only 43 cases were classified as being at ‘intermediate’ or ‘high’ risk for progressive disesase among 120 classifiable cases. Four other cases presented as liver metastasis. The overall patients’ mean age was 57.4 years (median: 60 years, range: 8–98 years; n=143), including five pediatric patients. Clinicopathological data were not available for all the patients, because most samples had been evaluated in the setting of a referral center with limited access to external clinical parameters.
Mutational Analysis
Two primary GISTs (cases 1 and 2) with an identical KIT exon 8 mutation (c.1255_1257delGAC) leading to the deletion of an aspartatic acid on residue 419 (p.D419del) were detected, representing 1.4% of all the cases investigated. The mutation was also found in a peritoneal metastasis of case 1, which was sequenced independently. No activating mutations in KIT exon 8 could be identified in the remaining 143 wild-type cases. However, silent mutations were found in KIT exon 17 (p.I798I, n=7, 4.8%) and PDGFRA exon 18 (p.V824V, n=24, 16.6%). Based on evaluation of the mutational status of 1351 well-characterized GIST samples from the Bonn–Cologne GIST registry, the overall frequency of KIT exon 8 mutations was 0.15%. In our referral center cohort, KIT exon 9 mutations were detected in 126 cases (9.33%), exon 11 mutations in 810 cases (59.96%), exon 13 mutations in 25 cases (1.85%), and exon 17 mutations in 23 cases (1.70%). PDGFRA mutations in exon 12 were found in 25 cases (1.85%), exon 14 mutations in 8 cases (0.59%), and exon 18 mutations in 189 cases (13.99%) (Figure 1).

Mutational status of 1351 GIST cases in the Bonn–Cologne GIST registry (n=1351), KIT mutations were found in 72.99% and PDGFRA mutations in 16.43% of cases. The remaining 10.58% were classified as wt-GISTs. The overall frequency of KIT exon 8 mutations is 0.15%.
Clinicopathological Characteristics of KIT Exon 8 Mutant GISTs
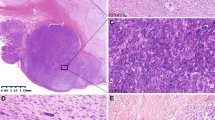
One GIST with a sporadic KIT exon 8 mutation (p.D419del) was found in a 53-year-old male patient. The tumor originated in the small bowel and measured 5.4 cm in diameter. The lesion showed a biphasic spindled-epithelioid histological appearance. Large tumor nodules consisted of epithelioid tumor cells with eosinophilic to clear cytoplasm, which were aggregated in an alveolar fashion. Multinucleated giant cells were not observed. High proliferative activity with numerous mitoses was noted and tumor necrosis was seen. The mitotic count was 13/50 HPF in this area. Furthermore, a second spindle cell pattern was noted. In this area, the mitotic count was 4/50 HPF (Figure 2, Table 2). According to the NCCN-AFIP criteria,28 the patient was classified as being at high risk of progressive disease. The patient developped multiple peritoneal metastases 29 months after surgical removal of the primary tumor. Mutational analysis of the metastasic lesions revealed the same deletion in KIT exon 8. Notably, a heterozygous mutation was detected in the primary tumor, whereas a homozygous pattern was detected in the metastatic disease. After 34 months, the patient was lost to follow up with progressive metastatic disease. As the patient was diagnosed in the pre-imatinib era, no adjuvant treatment was administered. Instead, the patient was treated with adriamycin and ifosfamide for his metastatic disease.

Histopathological and molecular features of a GIST with KIT exon 8 mutation. In case 1, epithelioid tumor cells with eosinophilic to clear cytoplasm were aggregated in an alveolar fashion ((a), × 200, H&E). A second tumor cell component showed a spindeled cell appearance and was situated in close proximity to the epithelioid areas ((b,c), × 200, H&E). Abundant necrosis was noted ((d), × 200, H&E). Metastatic tissue revealed epithelioid tumor cells ((e,f), × 200, H&E). Strong expression of the KIT receptor ((g), × 200, KIT). The p.D419del mutation occurred heterozygously in the primary tumor ((h), top) and homozygously in the metastasis ((h), bottom). FISH analysis showed polysomy by presenting multiple orange KIT signals as well as multiple green CEP4 signals in both the primary tumor and metastasis (i).
In case 2, the same heterozygous mutational subtype (p.D419del) was found in a GIST arising in the proximal jejunum. The 57-year-old female patient followed, however, a benign clinical course under adjuvant treatment (400 mg imatinib/day). The tumor measured 10 cm in size and a spindle cell phenotype of the entire tumor was observed (Figure 3, Table 2). One mitosis was found in 50 HPF. According to the NCCN-AFIP criteria,28 the patient was classified as being at moderate risk of progressive disease. Twenty-four months after removal of the GIST, there is no evidence of recurrence, while the patient is currently under adjuvant treatment with imatinib (400 mg/day).

Histopathological and molecular features of a GIST with KIT exon 8 mutation. In case 2, a spindled cell morphology was noted ((a), × 200, H&E). Immunohistochemistry revealed strong positivity for DOG1 (b, × 200) and KIT (c). Sanger sequencing showed a heterozygous deletion in KIT exon 8 (p.D419del) (d).
FISH Analysis
In case 1, the primary tumor showed the deletion in exon 8 of the KIT gene in a heterozygous pattern, whereas a homozygous status was detected in the relapse. In order to further elucidate the development of this homozygous pattern, we performed FISH analysis of the KIT gene locus in primary tumor and in the relapse. Spindle cell and epithelioid areas of the primary tumor did not differ in terms of KIT and centromer 4 copy numbers. Both the primary lesion and metastasis tumor cells showed polysomy by presenting multiple orange KIT signals as well as multiple green CEP4 signals (Figure 2). Thus, FISH analysis revealed neither a monosomy of chromosome 4 nor a mono-allelic deletion of the KIT gene locus as the cause for the homozygous mutational pattern.
Discussion
We have previously reported familial GISTs caused by germline mutation in KIT exon 8.24 This led us to the hypothesis that KIT exon 8 mutations might also represent driver mutations in a subset of sporadic GISTs. We therefore screened a well-defined cohort of 145 wt-GISTs for mutations in KIT exon 8.
On the molecular level, GISTs are characterized by mutations in the KIT or PDGFRA genes, both encoding for closely related tyrosine kinase proteins, which consist of regulatory and katalytic domains. The regulatory domain consists of an extracellular ligand-binding domain (EC) and a juxtamembrane domain (JM). Main parts of the katalytic domain are the two tyrosine kinase domains (TK1 and TK2), which are separated by a kinase insert.3 Each exon of KIT and PDGFRA corresponds to a specific molecular structure of the receptor. In sporadic GISTs, primary KIT mutations were hitherto found in EC and JM domains (exons 9 and 11, respectively) as well as in both kinase domains (exons 13 and 17); corresponding affected exons of PDGFRA are 12 (JM), 14 (TK1) and 18 (TK2).3 Beside GISTs, PDGFRA translocations or mutations are associated with various human neoplasms such as myeloproliferative diseases associated with hypereosinophilia29 or inflammatory fibroid polyps.25, 30 KIT mutations have also been reported in other malignancies, such as melanomas or seminomas.31 Some of these tumors share the same mutational hot spots and affected exons with GISTs.
KIT exon 8 mutations are located in the EC domain of the receptor and are frequently found in hematopoietic disorders, such as acute myeloid leukemia and/or mastocytosis.32, 33 Several mutational subtypes have been described that mostly affect codons 417–421 (Table 3). Up to now, KIT exon 8 mutations have not been systematically investigated in sporadic GISTs, and we report, for the first time, two cases with somatic mutations. In our cohort of wt-GISTs, which obviously appears to be representative, the frequency was 1.4%. However, they represent very rare mutational events, contributing to <0.2% of all GISTs.
In both our cases, we observed the same deletion (p.D419del), which was also shown to be associated with pediatric-onset cutaneous mastocytosis (urticaria pigmentosa) and acute myeloid leukemia (Table 3). The first case differed substantially from the second case in terms of morphology and clinical behavior: it had a peculiar biphasic and partially alveolar epithelioid growth pattern and followed an aggressive clinical course. A heterozygous mutation was found in the primary tumor, shifting to a homozygous mutation in the metastatic disease, which might reflect tumor progression. FISH analysis showed polysomy with more than three copies both of centromer 4 and the KIT locus in all the analyzed tumor cells. This finding rules out mono-allelic deletion of the wild-type KIT locus and is consistent with a duplication of the KIT mutant allele. This mechanism was previously described for KIT exon 11-mutated tumors by Lasota et al.34 They showed that a loss of heterozygosity was attributed to the loss of the wild-type allele with subsequent gain of one or multiple copies of the mutated one. As in our study, the phenomenon was associated with a malignant course of disease.
Our second case was an intermediate risk tumor of spindled subtype, which did not progress under adjuvant tyrosine kinase inhibition with imatinib. The 67-year-old female patient suffered from a small bowel tumor but is well with no evidence of disease 24 months after her surgery.
Exactly the same type of mutation was once reported as p.D419del germline mutation in familial GISTs. We could identify a kindred with both familiar GISTs and mastocytosis.24 These familial cases were typical low-risk GISTs with a spindled morphological subtype. By in vitro experiments, we could previously show that this particular mutation results in constitutively activated KIT, which can be inhibited by imatinib.24 In our present cases, no clue for neither a familiar background nor an associated mastocytosis was found, based on information from the patients’ files and oral consultation with their physicians.
We conclude that sporadic KIT exon 8 mutations in GISTs represent rare events. A small proportion (about 1–2%) of GISTs being formerly reported as ‘wild type’ might, in fact, carry a mutation in KIT exon 8. Only one mutational subtype (p.D419del) was identified so far that was shown to be activating and imatinib sensitive in vitro. We therefore propose that the screening for KIT exon 8 mutations should be implemented in the routine molecular diagnostics of GIST. In addition, patients with proven KIT exon 8 mutations and significant risk for tumor progression should be treated with imatinib.
References
Hirota S, Isozaki K, Moriyama Y et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580.
Heinrich MC, Corless CL, Duensing A et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710.
Lasota J, Miettinen M . Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008;53:245–266.
Miettinen M, Sobin LH, Lasota J . Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005;29:52–68.
Miettinen M, Makhlouf H, Sobin LH et al. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006;30:477–489.
Wardelmann E, Losen I, Hans V et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer 2003;106:887–895.
Lasota J, Dansonka-Mieszkowska A, Sobin LH et al. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest 2004;84:874–883.
Lasota J, Stachura J, Miettinen M . GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Invest 2006;86:94–100.
Antonescu CR, Sommer G, Sarran L et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003;9:3329–3337.
Corless CL, Schroeder A, Griffith D et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357–5364.
Debiec-Rychter M, Sciot R, Le Cesne A et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093–1103.
Heinrich MC, Owzar K, Corless CL et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360–5367.
Demetri GD, Benjamin RS, Blanke CD et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)—update of the NCCN clinical practice guidelines. J Natl Compr Cancer Netw 2007;5 (Suppl 2):S1–29 quiz S30.
Casali PG, Blay JY . Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21 (Suppl 5):v98–102.
Wardelmann E, Buttner R, Merkelbach-Bruse S et al. Mutation analysis of gastrointestinal stromal tumors: increasing significance for risk assessment and effective targeted therapy. Virchows Arch 2007;451:743–749.
Daniels M, Lurkin I, Pauli R et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett 2011;312:43–54.
Tarn C, Rink L, Merkel E et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci USA 2008;105:8387–8392.
Mussi C, Schildhaus HU, Gronchi A et al. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res 2008;14:4550–4555.
Miettinen M, Fetsch JF, Sobin LH et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006;30:90–96.
Carney JA, Stratakis CA . Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 2002;108:132–139.
Amar L, Bertherat J, Baudin E et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005;23:8812–8818.
Janeway KA, Kim SY, Lodish M et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA 2011;108:314–318.
Miettinen M, Lasota J, Sobin LH . Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005;29:1373–1381.
Hartmann K, Wardelmann E, Ma Y et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005;129:1042–1046.
Huss S, Wardelmann E, Goltz D et al. Activating PDGFRA mutations in inflammatory fibroid polyps occur in exons 12, 14 and 18 and are associated with tumour localization. Histopathology 2012;61:59–68.
Merkelbach-Bruse S, Dietmaier W, Fuzesi L et al. Pitfalls in mutational testing and reporting of common KIT and PDGFRA mutations in gastrointestinal stromal tumors. BMC Med Genet 2010;11:106.
Fassunke J, Blum MC, Schildhaus HU et al. qPCR in gastrointestinal stromal tumors: evaluation of reference genes and expression analysis of KIT and the alternative receptor tyrosine kinases FLT3, CSF1-R, PDGFRB, MET and AXL. BMC Mol Biol 2010;11:100.
Miettinen M, Lasota J . Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
Cools J, DeAngelo DJ, Gotlib J et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003;348:1201–1214.
Schildhaus HU, Cavlar T, Binot E et al. Inflammatory fibroid polyps harbour mutations in the platelet-derived growth factor receptor alpha (PDGFRA) gene. J Pathol 2008;216:176–182.
Went PT, Dirnhofer S, Bundi M et al. Prevalence of KIT expression in human tumors. J Clin Oncol 2004;22:4514–4522.
Beghini A, Larizza L, Cairoli R et al. c-kit activating mutations and mast cell proliferation in human leukemia. Blood 1998;92:701–702.
Lanternier F, Cohen-Akenine A, Palmerini F et al. Phenotypic and genotypic characteristics of mastocytosis according to the age of onset. PLoS One 2008;3:e1906.
Lasota J, vel Dobosz AJ, Wasag B et al. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab Invest 2007;87:1029–1041.
Shih LY, Liang DC, Huang CF et al. Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 2008;22:303–307.
Molderings GJ, Meis K, Kolck UW et al. Comparative analysis of mutation of tyrosine kinase kit in mast cells from patients with systemic mast cell activation syndrome and healthy subjects. Immunogenetics 2010;62:721–727.
Bodemer C, Hermine O, Palmerini F et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol 2010;130:804–815.
Markova J, Trnkova Z, Michkova P et al. Monitoring of minimal residual disease in patients with core binding factor acute myeloid leukemia and the impact of C-KIT, FLT3, and JAK2 mutations on clinical outcome. Leuk Lymphoma 2009;50:1448–1460.
Goemans BF, Zwaan CM, Cloos J et al. FLT3 and KIT mutated pediatric acute myeloid leukemia (AML) samples are sensitive in vitro to the tyrosine kinase inhibitor SU11657. Leuk Res 2010;34:1302–1307.
Beghini A, Ripamonti CB, Cairoli R et al. KIT activating mutations: incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica 2004;89:920–925.
Fuster O, Barragan E, Bolufer P et al. Rapid detection of KIT mutations in core-binding factor acute myeloid leukemia using high-resolution melting analysis. J Mol Diagn 2009;11:458–463.
Goemans BF, Zwaan CM, Miller M et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 2005;19:1536–1542.
Paschka P, Marcucci G, Ruppert AS et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol 2006;24:3904–3911.
Nanri T, Matsuno N, Kawakita T et al. Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t(8;21)(q22;q22). Leukemia 2005;19:1361–1366.
Gari M, Goodeve A, Wilson G et al. c-kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol 1999;105:894–900.
Park SH, Chi HS, Min SK et al. Prognostic impact of c-KIT mutations in core binding factor acute myeloid leukemia. Leuk Res 2011;35:1376–1383.
Ahmad F, D'Souza W, Mandava S et al. Molecular analysis of WT1 and KIT mutations in patients from an Indian population with de novo acute myeloid leukemia: determination of incidence, distribution patterns, and report of a novel KIT mutation. Leuk Lymphoma 2011;52:865–876.
Acknowledgements
This study was supported by a grant of the Manfred Stolte Foundation to H-US.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
SH, EW, SMB, and H-US received research grants and honoraria from Novartis Oncology. H-US received honoraria from Pfizer, Abbott Molecular, and Roche. All the other authors declare no conflict of interest.
Additional information
Parts of this work were presented at the 102nd annual meeting of the USCAP.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Huss, S., Künstlinger, H., Wardelmann, E. et al. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del). Mod Pathol 26, 1004–1012 (2013). https://doi.org/10.1038/modpathol.2013.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.47
Keywords
This article is cited by
-
New treatment strategies for advanced-stage gastrointestinal stromal tumours
Nature Reviews Clinical Oncology (2022)
-
Mesenchymale Tumoren und tumorartige Veränderungen des Gastrointestinaltraktes
Der Pathologe (2022)
-
Prevalence and significance of M541L single nucleotide polymorphism in the central European cohort of gastrointestinal stromal tumor patients
Journal of Cancer Research and Clinical Oncology (2021)
-
Low ETV1 mRNA expression is associated with recurrence in gastrointestinal stromal tumors
Scientific Reports (2020)
-
An update on adrenal endocrinology: significant discoveries in the last 10 years and where the field is heading in the next decade
Hormones (2018)
