
Abstract
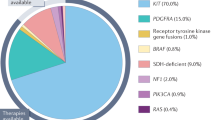
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the GI tract, arising from the interstitial cells of Cajal, primarily in the stomach and small intestine. They manifest a wide range of morphologies, from spindle cell to epithelioid, but are immunopositive for KIT (CD117) and/or DOG1 in essentially all cases. Although most tumors are localized at presentation, up to half will recur in the abdomen or spread to the liver. The growth of most GISTs is driven by oncogenic mutations in either of two receptor tyrosine kinases: KIT (75% of cases) or PDGFRA (10%). Treatment with tyrosine kinase inhibitors (TKIs) such as imatinib, sunitinib, and regorafenib is effective in controlling unresectable disease; however, drug resistance caused by secondary KIT or PDGFRA mutations eventually develops in 90% of cases. Adjuvant therapy with imatinib is commonly used to reduce the likelihood of disease recurrence after primary surgery, and for this reason assessing the prognosis of newly resected tumors is one of the most important roles for pathologists. Approximately 15% of GISTs are negative for mutations in KIT and PDGFRA. Recent studies of these so-called wild-type GISTs have uncovered a number of other oncogenic drivers, including mutations in neurofibromatosis type I, RAS genes, BRAF, and subunits of the succinate dehydrogenase complex. Routine genotyping is strongly recommended for optimal management of GISTs, as the type and dose of TKI used for treatment is dependent on the mutation identified.
Similar content being viewed by others
Main
Gastrointestinal stromal tumors (GISTs) occur primarily in older patients of either sex, with annual incidences between 11 and 19.6 per million population worldwide.1, 2, 3, 4 This corresponds to between 3300 and 6000 new cases per year in the United States. Following surgical resection, GISTs often recur locally, spread diffusely throughout the serosal surfaces of the abdomen, and/or metastasize to the liver. Advanced disease is associated with metastases to distant sites, including the lung and bone. Before the advent of targeted therapies, the prognosis for advanced GISTs was poor owing to their inherent resistance to both chemotherapy and radiation therapy.
During the past decade, GISTs have served as an important model in the emerging field of molecularly targeted therapies for solid tumors. The nearly simultaneous discovery of oncogenic kinase mutations in GISTs and the introduction of kinase inhibitors have led to a rapid evolution in our understanding of these tumors and the biology that defines them.
Clinical and pathological features
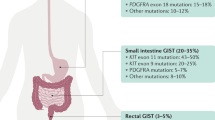
GISTs are most commonly present in the stomach (60%) and small intestine (25%), but they also arise in the colon, rectum, esophagus, mesentery, and omentum (15% together).5, 6 Clinical symptoms associated with GISTs include fatigue, abdominal pain, dysphagia, satiety, and obstruction. The workup often reveals anemia related to mucosal bleeding or intratumoral hemorrhage.
The tumors are generally well circumscribed, have a fleshy pink or tan cut surface, and may show areas of hemorrhagic necrosis and cystic degeneration. They range from 1 cm to more than 40 cm, with an average of ∼5 cm. Morphologically, GISTs show a wide spectrum of morphologies, from bland spindle cell proliferations to highly cellular epithelioid tumors with significant nuclear pleomorphism (Figure 1). For these reasons, the morphologic differential diagnosis is necessarily broad (Table 1). Skeinoid fibers are associated with lower-grade lesions as are foci of calcification.

Examples of gastrointestinal stromal tumor (GIST) morphology. Spindle cell GISTs (left panels) often harbor a mutation in KIT exon 9 or 11. Epithelioid GISTs (right panels) vary in their genotype, having either (or neither) a KIT or PDGFRA mutation.
CD34 is expressed in 70% of GISTs and was the first immunohistochemical marker that helped to distinguish these tumors from leiomyomas and leiomyosarcomas of the GI tract. In 1998, reports by two groups that GISTs commonly express KIT (CD117) led to more reliable diagnosis.7,8 The staining may be membranous, diffusely cytoplasmic, or concentrated in a dot-like perinuclear pattern. The addition of DOG1 (ANO1) as another GIST marker has made the diagnosis quite routine,9 as DOG1 and CD117 each stain >95% of GISTs and, between them, serve to mark essentially all cases. These antigens are only rarely expressed in other mesenchymal tumors. Smooth muscle actin and muscle-specific actin are variably expressed in GISTs, whereas desmin is usually absent. Immunostaining for SDHA and SDHB has recently been shown to be effective in identifying tumors deficient in succinate dehydrogenase activity,10 the significance of which is discussed below.
Oncogenic mutations in GISTs
KIT
In 1998, Hirota et al7, 8 published their breakthrough discovery of KIT mutations in GISTs. Approximately 75% of GISTs harbor a KIT gene mutation, and these mutations lead to constitutive activation of the kinase. KIT is a member of the type III receptor tyrosine kinase family that includes platelet-derived growth factor receptors-α and -β (PDGFRA and PDGFRB), as well as the macrophage colony-stimulating-factor receptor (CSF1R) and the Fl cytokine receptor (FLT3).11 Binding of the KIT ligand (stem cell factor, SCF) to KIT results in receptor homodimerization and kinase activation.
Oncogenic KIT mutations cause ligand-independent kinase activation. The most common mutations in KIT affect the juxtamembrane domain encoded by exon 11 (Figure 2). Two-thirds of GISTs harbor mutations in exon 11 that disrupt the normal juxtamembrane secondary structure that keeps the kinase activation loop from swinging into an active conformation. These mutations include in-frame deletions, insertions, substitutions, or combinations thereof. The deletions are associated with a shorter progression-free and overall survival in comparison with the other exon 11 mutations.12, 13, 14, 15, 16, 17, 18 In particular, deletions involving codons 557 and/or 558 are associated with malignant behavior.19, 20, 21

KIT and PDGFRA mutations in gastrointestinal stromal tumor (GIST).
Aside from exon 11 mutations, between 7 and 10% of GISTs have a mutation in an extracellular domain encoded by exon 9.22 These mutations are thought to mimic the conformational change that the extracellular KIT receptor undergoes when ligand is bound. Importantly, the kinase domain in exon 9-mutant KIT is essentially the same as in wild-type KIT, and this influences inhibitor sensitivity. Interestingly, these mutations occur in tumors arising in the small and large intestine, but are very rarely seen in gastric GISTs.
Mutations in the activation loop (encoded by exon 17) of the kinase are uncommon. They stabilize the active conformation.23 Primary mutations, such as K642E in the ATP-binding region (encoded by exon 13), are also uncommon.23 The biological basis of kinase activation by this mutation is unknown, but it is speculated that it interferes with normal auto-inhibitory function of the juxtamembrane domain.
The functional importance of KIT mutations in GIST development is supported by several lines of evidence. First, phosphorylated (activated) KIT is detectable in GIST tumor extracts. Second, mutant KIT is oncogenic, supporting the growth of stably transfected BA/F3 cells in nude mice.7, 24 Third, when expressed in transfected cell lines, mutant forms of KIT show constitutive kinase activity in the absence of SCF, as evidenced by autophosphorylation and activation of downstream signaling pathways.7, 25 Finally, mice engineered to express KIT with mutations of the type found in human GISTs develop diffuse ICC hyperplasia within the muscular wall of the stomach and intestine.26, 27 These mice also develop GIST-like tumors. This histologic picture is similar to that seen in individuals with inherited KIT-activating mutations.28, 29
Tumor extracts from KIT-mutant GISTs demonstrate evidence of activation of downstream signaling pathways, including the MAP kinase pathway (RAF, MEK, and ERK), the PI3 kinase/AKT pathway, and STAT3 (Figure 3a).24, 30, 31, 32 The MAP kinase pathway upregulates important transcriptional regulators such as MYC, ELK, and CREB, and can stimulate the cell cycle through FOS. AKT activation through PI3 kinase and PDK1 leads to increased protein translation, downregulation of the cell cycle inhibitor p27KIP, and anti-apoptotic effects. Recent studies have shown that ETV1 is an important regulator of GIST-specific gene expression during tumorigenesis.33 KIT signaling through the MAP kinase pathway serves to maintain ETV1 activity.

(a) KIT and PDGFRA cell signaling pathways. Dimerization of KIT or platelet-derived growth factor receptors-α (PDGFRA) leads to signaling through the mitogen-activated protein (MAP) kinase pathway (RAF, MEK and ERK) and the phosphoinositide 3-kinase (PI3K) pathway (AKT, mammalian target of rapamycin (mTOR), S6 kinase). In addition, signal transducer and activator of transcription 3 (STAT3) is activated. The collective impact favors an increase in cell metabolism, cell cycle progression, and a decreased sensitivity to apoptosis. (b) Cell signaling in ‘wild-type’ gastrointestinal stromal tumors (GISTs). Mutations in neurofibromatosis type I (NF1), RAS genes, or BRAF lead to increased signaling through the MAP kinases MEK and ERK, promoting cell growth. Loss of the mitochondrial succinate dehydrogenase complex through mutations in SDHA, SDHB, SDHC, or SDHD, leads to an accumulation of succinate, which inhibits prolyl hydroxylase-mediated degradation of hypoxia-inducible factor-1α (HIF1α) through the proteosome complex. This results in upregulated transcription of a number of genes, including VEGF, IGF1, and IGF2. Succinate also inhibits demethylation of DNA by TET2 through increasing α-ketoglutarate levels.
In general, GISTs are heterozygous for a given mutation; however, in ∼15% of tumors the remaining wild-type KIT allele is lost and this is associated with malignant behavior.29, 34, 35, 36 In serial samples from individual patients, Chen et al36 have provided evidence that this occurs through mitotic non-dysjunction, ie, failure of a chromosome 4 pair bearing the wild-type KIT allele to separate during mitosis, leaving one daughter cell with a single chromosome 4 containing the mutant KIT allele (uniparental monosomy). This correlates with increased mitotic activity and topoisomerase II expression.
Platelet-Derived Growth Factor Receptor-α
Immunoblots of GIST extracts from tumors lacking KIT gene mutations sometimes show high levels of phosphorylation of the α-receptor for platelet derived growth factor (PDGFRA), which is a close homolog of KIT.37 PDGFRA is activated in GISTs harboring mutations in the juxtamembrane domain (exon 12), the ATP-binding domain (exon 14), or the activation loop (exon 18) (Figure 2 and Table 2). Consistent with their extensive functional overlap, KIT and PDGFRA mutations are mutually exclusive in GISTs.
Observations supporting the significance of PDGFRA mutations in GIST parallel to those for KIT mutations. When expressed in transfected cell lines, mutant forms of PDGFRA have constitutive kinase activity in the absence of their ligand, PDGF-A,37, 38 the activated downstream pathways are identical to those in KIT-mutant GISTs,37, 39 and PDGFRA is also stabilized by HSP90.40 In addition, both types of tumors are immunopositive for the markers DOG1 and protein kinase C-θ (PKCθ).9, 41, 42 These markers are highly selective for GISTs over other mesenchymal tumors. Further, as discussed below, both genotypes are associated with cytogenetic changes that are distinctive for GIST.37, 43
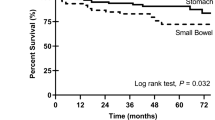
Despite these molecular similarities, PDGFRA-mutant GISTs do show features distinctive from KIT-mutant GISTs, including differences in gene expression profile,39, 44 a striking predilection for the stomach, variable (sometimes negative) expression of KIT,20, 41, 45, 46, 47, 48 and a generally lower potential for malignancy.49 Morphologically, however, PDGFRA-mutant GISTs are not reliably distinguishable from KIT-mutant GISTs (Figure 1).
Other Driver Mutations
Approximately 15% of GISTs do not have a detectable mutation in either KIT or PDGFRA. In other respects, these so-called ‘wild-type’ GISTs are clinically indistinguishable from KIT- or PDGFRA-mutant GISTs, having identical morphology, expressing high levels of KIT, and occurring anywhere in the GI tract. Phosphorylated KIT is detectable in these tumors, suggesting that KIT is still activated,30 but the mechanism of this activation is unclear. However, recent studies have revealed that wild-type GISTs are a heterogeneous group and display various oncogenic mutations (Table 2). For example, the BRAF V600E substitution common in papillary thyroid carcinoma and melanoma is present in up to 13% of wild-type GISTs.50 HRAS, NRAS and PIK3CA gene mutations also occur, but are much more rare. As BRAF and the RAS proteins are constituents of the MAP kinase signaling cascade, they can result in KIT-independent growth stimulation (Figure 3b) and are possible causes for resistance to KIT/PDGFRA kinase inhibitors.
It is estimated that 7% of patients with neurofibromatosis type I (NF1) develop one or more GISTs.51, 52, 53, 54, 55, 56 Most arise in the small intestine and they do not readily metastasize.51 The majority of these GISTs are wild type for KIT and PDGFRA, but as expected they do show either somatic mutation or loss of the remaining wild-type NF1 allele, resulting in signaling through the MAP kinase cascade (Figure 3b).51, 54, 55, 57
SDH-Deficient GISTs
Approximately half of all wild-type GISTs show loss of respiratory chain complex II enzymatic activity.10, 58 This complex comprises four subunits (SDHA, SDHB, SDHC, and SDHD) and serves to oxidize succinate to fumarate as part of the mitochondrial Krebs cycle. Loss of any of these subunits through gene mutation or post-transcriptional downregulation destabilizes the complex and causes the accumulation of succinate. This results in increased transcription of HIF1α-regulated genes and decreased DNA demethylation (Figure 3b).59 Indeed, SDH-deficient GISTs show a global increase in DNA methylation similar to that seen in gliomas and leukemias with IDH1 and IDH2 mutations.60
Germline mutations in SDHA, SDHB, or SDHC increase the risk not only for the development of one or more SDH-deficient GISTs but also for paragangliomas (Carney–Stratakis syndrome).10, 61 The GISTs in affected patients show either loss or somatic mutation (second hit) of the remaining wild-type allele. The absence of immunostaining for SDHA, as well as SDHB, is helpful in identifying GISTs with possible SDH gene mutations.58, 58, 62, 63, 64, 65, 66
Approximately 50% of wild-type GISTs demonstrate high expression of IGF1R.67 In SDH-deficient GISTs, upregulation of IGF1 and IGF2 may activate IGF1R in an autocrine manner (Figure 3b), resulting in signaling through both the MAP kinase and PI3 kinase/AKT pathways.68
GISTs that arise in pediatric patients (∼1–2% of all GISTs) are predominantly SDHB immunonegative, but rarely harbor an SDH gene mutation. These tumors, which often metastasize but tend to grow slowly, have a different gene expression signature from KIT/PDGFRA-mutant GISTs.69, 70, 71 Some pediatric-type GISTs are accompanied by pulmonary chondromas and/or paragangliomas, referred to as Carney triad, a non-heritable syndrome, the genetic cause for which has yet to be determined.72
The origin of GISTs
Interstitial Cells Of Cajal
During the 1990s, a number of investigators noted similarities between GISTs and a population of cells in the GI tract called the interstitial cells of Cajal (ICCs), which serve as pacemakers for peristaltic contractions. These observations led to the hypothesis that ICCs could be the cell-of-origin of GISTs. Mice engineered to express KIT with mutations of the type found in human GISTs develop diffuse ICC hyperplasia within the muscular wall of the stomach and intestine.26, 27 These mice also develop GIST-like tumors. Diffuse ICC hyperplasia has been described in several kindreds with heritable mutations in KIT (Table 2), and is associated with dysphagia and the development of multiple GISTs,26, 29, 53, 73, 74, 75, 76, 77, 78 although many of the tumors do not follow a malignant course.
The relationship between GISTs and ICCs is further supported by parallels in gene expression. For example, high levels of PKCθ, nestin, and DOG1 are expressed in both GISTs and ICCs. In addition, the ETS family transcription factor ETV1 is highly expressed in both GISTs and in the specific subpopulations of ICCs (myenteric and intramuscular, as opposed to submucosal) thought to give rise to GISTs.33
The observation that some KIT and PDGFRA mutations in GISTs correlate closely with anatomical location (Table 2) might be explained by their ICC origin. For example, GISTs with a KIT exon 9 mutation, which arise primarily in the intestines, may derive from a different subgroup of ICCs than those with a PDGFRA D842V mutation, which occur only in the stomach, mesentery, and omentum. The more common KIT mutations, by contrast, can be found in GISTs throughout the GI tract, perhaps deriving from a more ubiquitous ICC subtype.
Micro-GISTs
Minute growths (1–10 mm) of ICC/GIST-like cells are present in between 2.9 and 35% of the stomachs thoroughly examined after surgical removal or at autopsy.79, 80, 81, 82 These so-called micro-GISTs are mitotically inactive and often partially calcified, suggesting tumorigenic arrest. In contrast to the diffuse ICC hyperplasia observed in the presence of a germline KIT mutation, micro-GISTs appear to represent a nodular form of ICC overgrowth caused in most cases by local, somatic acquisition of a KIT mutation. The type and frequency of KIT mutations in micro-GISTs is essentially the same as in clinically significant tumors.83 Subcentimeter GISTs with PDGFRA mutations have also been reported.79 These observations on micro-GISTs suggest that kinase gene mutations occur very early in GIST tumorigenesis; however, the mutations are probably not sufficient for progression to an oncologically threatening lesion (Figure 4). The large pool of micro-GISTs in the general population likely explains the multiple reported cases in which two or more genotypically distinct GISTs are found in a patient during a single surgical procedure.53, 79, 84

Origin and progression of gastrointestinal stromal tumors (GISTs). Under the influence of a KIT or platelet-derived growth factor receptors-α (PDGFRA) mutation, a clonal outgrowth of interstitial cells of Cajal (ICCs) may form a discrete nodule (micro-GIST) measuring <1 cm and showing essentially no mitotic activity. A conservative estimate for the prevalence of micro-GISTs in the US population would exceed 10 million lesions. With the accumulation of additional mutations and chromosomal changes, a micro-GIST may progress to a larger lesion that is clinically significant. Approximately 5000 such lesions are identified each year in the United States, of which 2000 are of high risk or are malignant.
Chromosomal and Molecular Alterations During GIST Progression
Although oncogenic kinase mutations have a significant role in the development of GISTs, other genetic events are important in their clinical progression (Figure 4). Approximately two-thirds of GISTs demonstrate either monosomy of chromosome 14, or partial loss of 14q.37, 85, 86, 87, 88, 89 Interestingly, these chromosome 14 abnormalities are observed in both KIT-mutant and PDGFRA-mutant GISTs.37, 43 Deletions of 14q11.2 include the genes PARP2, APEX1, and NDRG2, whereas deletions of 14q32 include the SIVA gene.90 Loss of the long arm of chromosome 22 is observed in ∼50% of GISTs.37, 43, 85, 86, 89, 91
Losses on chromosomes 1p, 9p, 11p, and 17p are successively less common than 14q and 22q losses, but are more significantly associated with malignancy (Figure 4).37, 43, 85, 89, 91, 92, 93, 94 Losses on chromosomes 10, 13q, and 15q have also been reported in GISTs.43, 91 Gains on chromosomes 8q (including MYC), 3q (including SMARCA3), and 17q are associated with metastatic behavior.86, 95
In a recent array-based analysis of gene copy number in 42 GISTs (23 with recurrence or metastasis), the tumors were separated into four groups reflecting their accumulated chromosomal changes. The overall survival of group 1 (loss of 22q, 19, and 1p distal) and group 2 (additional loss of 14q) was significantly better than that of group 3 (added losses of 15q and 1p proximal) and group 4 (additional loss of 10). Specific genes implicated in this analysis included OXA1L on 14q, as well as AKAP13 and C15orf5 on 15q.
None of the above karyotypic changes is present in pediatric-type GISTs, which remain near-diploid, again emphasizing the different biology of these tumors. In contrast, GISTs arising in NF1 patients often show losses of 14q and 22q.96
On the basis of gene expression profiling of high-risk versus low-risk GISTs, the high-risk tumors show significant changes in genes regulating the cell cycle, including genes influenced by the PI3 kinase pathway and genes involved in the G2/M checkpoint.97 A significant fraction of malignant GISTs show inactivation of the tumor suppressor gene CDKN2A (which encodes the cell cycle regulatory protein p16INK4A) through chromosome 9p21 deletion, either biallelic or in combination with mutation or promoter methylation.98, 99, 100, 101 TP53 mutations and decreased p53 immunostaining also correlate with a poor prognosis.102, 103, 104 Likewise, amplifications of MDM2 and CCND1 (cyclin D1), although uncommon in GISTs, are associated with malignancy.105
Kinase mutations and tyrosine kinase inhibitor therapy
Until the year 2000, treatment options for patients with advanced GIST were poor. The response rate to conventional chemotherapy was <5% and median survival for patients with advanced disease was ∼18 months.
The tyrosine kinase inhibitor (TKI) imatinib was developed in the early 1990s as a treatment for chronic myelogenous leukemia because of its ability to inhibit the fusion oncoprotein BCR–ABL.106 The observation that ABL shares structural similarity with KIT, and several other tyrosine kinases led to experiments showing that imatinib can inhibit the growth of cells expressing mutant forms of KIT.25 In addition, imatinib showed potent activity against a KIT-mutant GIST cell line. Imatinib inhibits KIT kinase by directly binding to the ATP pocket and competitively inhibiting ATP binding. The KIT receptor is normally in equilibrium between active and inactive conformations. The latter is favored by steric hindrance conferred by the juxtamembrane domain (exon 11), which prevents the activation loop from assuming the conformation required for kinase activation.
With the knowledge that imatinib inhibits KIT signaling, imatinib was first used clinically to treat a 50-year-old female with metastatic GIST, and a dramatic response was observed.107 Promising results from phase I and II trials led to two international phase III trials, each using similar protocols that would allow a subsequent meta-analysis. The phase III trials compared 400 versus 800 mg daily doses of imatinib. Overall, imatinib achieved disease control in 70–85% of patients with advanced KIT-immunopositive GISTs, and the median progression-free survival was 20–24 months.108, 109, 110, 111, 112 Currently, the median survival for patients with advanced disease treated with frontline imatinib is 5 years, with 34% of patients surviving more than 9 years.113
A meta-analysis of the phase III trials proved that patients with exon 9-mutant GIST had a significantly longer progression-free survival if they were treated with 800 mg of imatinib; hence, this is now the target dose for these patients. In contrast, only 400 mg of imatinib is needed to manage exon 11-mutant tumors.112
The success with imatinib in patients with metastatic disease quickly led to trials of the drug in the adjuvant setting. It was initially shown that 12 months of imatinib after primary resection of a GIST significantly delayed disease recurrence versus a placebo.114 A subsequent trial proved that 36 months of adjuvant imatinib was superior to 12 months.115
Assessing GIST prognosis
FDA approval for the use of imatinib in the adjuvant setting has made it imperative that the prognosis of a newly resected GIST be predicted as accurately as possible. In a 2-year prospective study of sarcomas in France, >85% of GISTs presented as localized lesions.116 Thus, the question of whether to use adjuvant therapy must be considered for the great majority of newly diagnosed GISTs. Kinase genotype does not factor into overall survival once a GIST becomes metastatic. Thus, a KIT or PDGFRA mutation may set the initial course of a GIST, but the prognosis at the time of clinical presentation is clearly influenced by other genetic events. Unfortunately, our knowledge of these additional mutations remains limited, and current recommendations for assessing prognosis are based on three simple parameters: tumor size, tumor location, and mitotic index (mitoses/5 mm2). A number of risk assessment schemes have been published; the most widely used scheme was developed at the Armed Forces Institute of Pathology by Miettinen et al117, 118, 119, whose considerable efforts in studying the outcome of patients before the advent TKIs have provided the most complete data available (Table 3). It should be noted that tumor rupture, either before or during surgery, is another important negative prognostic factor. Incomplete resection, particularly in the area of the rectum, is also associated with a higher risk of recurrence.120
As a group, PDGFRA-mutant GISTs appear to be less aggressive than KIT-mutant GISTs,49 yet PDGFRA-mutant tumors can still progress and kill patients. These tumors are assessed using the same criteria as other GISTs.
Responses to TKI therapy
Clinical Disease Persistence
Clinical data suggest that even long-term TKI treatment fails to eradicate GIST cells, resulting in disease persistence. In an attempt to determine the optimal duration of imatinib therapy for advanced, unresectable GIST, an interesting trial randomized patients who had continuous control of their disease during 3 years of imatinib treatment to either continue or to discontinue treatment.121 For those continuing treatment progression-free survival over the next 2 years was 80%, but for those who stopped therapy it was only 16%. Patients who relapsed after discontinuation of therapy did so because of persistent disease. In contrast, the progression that developed in some of the patients who maintained therapy was due to resistant disease.
Persistence of GIST cells during TKI treatment could be due to the failure of these drugs to eradicate mature GIST cells and/or the failure to eliminate GIST stem cells. Current evidence suggests that both mechanisms underlie GIST persistence in the face of prolonged TKI therapy. Agaram et al122 examined a series of 43 clinically responsive GISTs lesions from 28 patients. Histological responses in these resected tumors after 1–31 months of imatinib treatment ranged from <10% to >90% reduction in tumor cellularity, but tumor cells persisted in all lesions. Three quarters of the lesions showed an absence of mitoses and a proliferative index of 0% by Ki-67 staining, indicating biological quiescence. Interestingly, some of the tumor cells showed transdifferentiation. Indeed, examples of rhabdomyoblastic, cartilaginous, and osseous differentiation have all been observed in cases of treated GIST (Figure 5). Thus, under imatinib suppression, GIST cells may avoid apoptosis by exiting the cell cycle and expressing genes associated with a differentiated phenotype. Whether these cells proliferate again when a TKI is discontinued, or whether GIST stem cells are the source of renewed growth, has not been determined.

Differentiation of gastrointestinal stromal tumor (GIST) cells under suppression of a tyrosine kinase inhibitor (TKI). In some cases, imatinib treatment may lead to rhabdomyoblastic differentiation (left panel), or else chondroid or osseous differentiation (right panels).
In rare cases, GISTs may progress to high grade, anaplastic sarcomas that lose CD117 expression. This has been observed in both imatinib-treated and TKI-naive tumors.123
Resistance to TKI therapy
Primary Resistance
Resistance to treatment with KIT/PDGFRA inhibitors such as imatinib can be divided into two types: primary and secondary. Approximately 10% of patients with GIST have primary resistance, defined as progression within the first 6 months of treatment. On the basis of data from phase II and phase III trials, tumor response to imatinib correlates with the underlying kinase genotype.34, 41, 112, 124, 125 The probability of primary resistance to imatinib for KIT exon 11, KIT exon 9, and wild-type GISTs is 5, 16 and 23%, respectively.34 These findings likely reflect the underlying sensitivities of different KIT genotypes. Exon 11-mutant KIT is highly sensitive to imatinib, with an IC50 of <100 nM, whereas exon 9-mutant KIT and wild-type KIT are less sensitive to the drug (IC50 ∼1000 nM for each).126 Thus, underdosing of imatinib in patients with exon 9 mutations probably accounts for some of the apparent resistance.112
On the basis of in vitro data, the most common PDGFRA mutation in GISTs, D842V, is fully resistant to the effects of imatinib.34, 38, 127, 128 This mutation favors the active conformation of the kinase domain and consequently disfavors imatinib binding.34, 129, 130 This is corroborated by clinical results, as patients with PDGFRA D842V-mutant GIST have low response rates and very short progression-free and overall survivals during imatinib treatment. Crenolanib is a TKI that has activity versus D842V and is now being tested in a clinical trial. There are, however, other PDGFRA mutations that are sensitive to imatinib in vitro and patients with these mutations have shown durable responses to imatinib.
Wild-type GISTs include tumors with mutations downstream of KIT;10, 50, 131, 132 hence, these subsets of wild-type GISTs might respond better to other targeted agents, such as VEGFR inhibitors for pediatric/SDH-deficient GIST and BRAF/MEK inhibitors for BRAF and RAS mutant GIST.133
Secondary Resistance
After an initial response to imatinib, the vast majority of patients eventually develop disease progression. This secondary resistance may manifest in a number ways, including growth of a nodule within a pre-existing, clinically quiescent lesion, the development of one or more new lesions, or widespread disease throughout the liver or abdominal cavity. It is now established that acquired mutations in KIT or PDGFRA account for most secondary resistance, and that these mutations occur almost exclusively in the same gene and allele as the primary oncogenic driver mutation.35, 134, 135, 136, 137, 138, 139, 140
In a phase II imatinib study for advanced GIST, 67% of the patients whose tumor showed imatinib resistance had a new, or secondary, mutation in KIT. Notably, these mutations were common among tumors with a primary exon 11 mutation, but were not observed in wild-type GIST samples.137 Unlike primary mutations that activate KIT, which are predominantly in the juxtamembrane regions encoded by exons 9 and 11, the secondary mutations were concentrated in two regions of the KIT kinase domain. One is the ATP-binding pocket, encoded by exons 13 and 14, mutations of which directly interfere with drug binding. The second is the activation loop (exons 17 and 18), where mutations can stabilize KIT in the active conformation and thereby hinder drug interaction. Drug resistance has also been observed in PDGFRA-mutant GISTs, in which the most common one is an acquired D842V mutation (activation loop).135, 137
Additional studies using more sensitive assays have identified secondary mutations in >80% of drug-resistant GIST lesions.141, 142, 143 More sobering is that there is significant heterogeneity of resistance across different lesions, and even within different areas of the same lesion. For example, there are reports of up to five different drug resistance mutations in different portions of an individual lesion and up to seven different secondary resistance mutations across multiple tumors in the same patient.141 This heterogeneity of resistance significantly impacts the efficacy of salvage TKI therapy after frontline imatinib, because the diversity of resistant, minority clones precludes the systemic suppression of GIST cells by any particular TKI.
Approaches to imatinib-resistant GIST
Most GIST patients who develop secondary resistance to imatinib will not respond to dose escalation, forcing a switch to an alternative KIT/PDGFRA TKI. Such salvage agents include sunitinib, sorafenib, regorafenib, vatalanib, masatinib, nilotinib, and dasatinib, as well as other investigational inhibitors. Although all of these agents are KIT/PDGFRA inhibitors, the majority of them (in contrast to imatinib) also target VEGFR1/2.144 Whether the disease stabilization that can be seen with these salvage agents is related to VEGFR1/2 inhibition and a consequent decrease in angiogenesis remains unclear.
Sunitinib is FDA-approved for the treatment of GIST patients with progression on imatinib,145 but biochemical evidence suggests that its activity against secondary imatinib-resistant kinase mutations is suboptimal. KIT ATP-binding pocket mutations are extremely sensitive to sunitinib in vitro; however, the activation loop mutations are strongly cross-resistant. Given the approximately equal frequency of these different classes of mutations in imatinib-resistant lesions and the multiplicity of lesions in a typical patient, it is not surprising that mixed responses are common during sunitinib therapy.124, 146
Most recently, the FDA has approved regorafenib for the third-line treatment of patients with GIST that is resistant to imatinib and sunitinib. In a phase III trial, the progression-free survival on regorafenib was 4.8 months.147 Thus, even with newer drugs such as regorafenib, resistance develops over time, suggesting that escape from ATP-competitive inhibitors of KIT and PDGFRA is inevitable. A new class of non-ATP mimetic kinase inhibitors (switch pocket kinase inhibitors, such as DP-2976) have shown high potency when tested in vitro and represent hope in the fight against TKI resistance.148, 149
Summary and future directions
Achievements in the treatment of GISTs during the past decade are the direct result of a growing understanding of their molecular biology. The high frequency of primary KIT and PDGFRA mutations in these tumors makes them sensitive to kinase inhibitors such as imatinib, and this drug is FDA-approved for use in both the adjuvant and advanced disease settings. Accurate assessment of the risk of disease recurrence is essential in determining the use of adjuvant imatinib after resection of a primary tumor. Current recommendations for assessing recurrence risk are based on tumor size, tumor location, and mitotic index (mitoses/5 mm2).117, 118, 119
Resistance to imatinib develops in the majority of advanced cases of GIST. An immediate research goal is the development of new inhibitors that have activity against secondary mutations in the activation loop. In addition, the development of effective combination therapy is likely to improve tumor control. Ongoing high-throughput genomic studies may identify additional drivers/modifiers of GIST biology that can be targeted.
Clinical genotyping of GISTs is important to identify KIT exon 9-mutant tumors that require a higher dose of imatinib for optimal disease control. Further, there are other molecular subtypes of GIST that do not respond well to conventional KIT inhibitors, but may be better treated with other agents (eg, SDH-deficient GIST and PDGFRA D842V GIST). Thus, the GIST genotyping is critical in personalizing the care of patients who need TKI therapy.
In summary, new insights into the origin and progression of GISTs are setting the stage for further therapeutic innovations, with the goal not just to control disease growth, but to eliminate all tumor cells at the time of initial therapy. Going forward, the challenge will be to move from a paradigm of tumor suppression to true cancer cure.
References
Chan KH, Chan CW, Chow WH et al. Gastrointestinal stromal tumors in a cohort of Chinese patients in Hong Kong. World J Gastroenterol 2006;12:2223–2228.
Goettsch WG, Bos SD, Breekveldt-Postma N et al. Incidence of gastrointestinal stromal tumours is underestimated: Results of a nation-wide study. Eur J Cancer 2005;41:2868–2872.
Nilsson B, Bumming P, Meis-Kindblom JM et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era. Cancer 2005;103:821–829.
Tryggvason G, Gislason HG, Magnusson MK et al. Gastrointestinal stromal tumors in Iceland, 1990-2003: the Icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer 2005;117:289–293.
Corless CL, Fletcher JA, Heinrich MC . Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813–3825.
Miettinen M, Lasota J . Gastrointestinal stromal tumors (GISTs): definition, occurrence, pathology, differential diagnosis and molecular genetics. Pol J Pathol 2003;54:3–24.
Hirota S, Isozaki K, Moriyama Y et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580.
Kindblom LG, Remotti HE, Aldenborg F et al. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998;152:1259–1269.
West RB, Corless CL, Chen X et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 2004;165:107–113.
Janeway KA, Kim SY, Lodish M et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA 2011;108:314–318.
Hanks SK, Quinn AM, Hunter T . The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988;241:42–52.
Ernst SI, Hubbs AE, Przygodzki RM et al. KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998;78:1633–1636.
Lasota J, Jasinski M, Sarlomo-Rikala M et al. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999;154:53–60.
Singer S, Rubin BP, Lux ML et al. Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol 2002;20:3898–3905.
Taniguchi M, Nishida T, Hirota S et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999;59:4297–4300.
Andersson J, Bumming P, Meis-Kindblom JM et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology 2006;130:1573–1581.
Cho S, Kitadai Y, Yoshida S et al. Deletion of the KIT gene is associated with liver metastasis and poor prognosis in patients with gastrointestinal stromal tumor in the stomach. Int J Oncol 2006;28:1361–1367.
Liu XH, Bai CG, Xie Q et al. Prognostic value of KIT mutation in gastrointestinal stromal tumors. World J Gastroenterol 2005;11:3948–3952.
Martin J, Poveda A, Llombart-Bosch A et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol 2005;23:6190–6198.
Tzen CY, Mau BL . Analysis of CD117-negative gastrointestinal stromal tumors. World J Gastroenterol 2005;11:1052–1055.
Wardelmann E, Losen I, Hans V et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer 2003;106:887–895.
Lux ML, Rubin BP, Biase TL et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000;156:791–795.
Lasota J, Corless CL, Heinrich MC et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol 2008;21:476–484.
Rubin BP, Singer S, Tsao C et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001;61:8118–8121.
Heinrich MC, Griffith DJ, Druker BJ et al. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000;96:925–932.
Sommer G, Agosti V, Ehlers I et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci USA 2003;100:6706–6711.
Rubin BP, Antonescu CR, Scott-Browne JP et al. A knock-in mouse model of gastrointestinal stromal tumor harboring kit K641E. Cancer Res 2005;65:6631–6639.
Chen H, Hirota S, Isozaki K et al. Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut 2002;51:793–796.
O'riain C, Corless CL, Heinrich MC et al. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol 2005;29:1680–1683.
Duensing A, Medeiros F, McConarty B et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 2004;23:3999–4006.
Rossi F, Ehlers I, Agosti V et al. Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci USA 2006;103:12843–12848.
Duensing A, Joseph N, Liu C et al. Protein kinase C theta (PKC{theta}) is selectively expressed and constitutively phosphorylatedd in gastrointestinal stromal tumors (GISTs). AACR Meet Abstr 2004;2004:708.
Chi P, Chen Y, Zhang L et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 2010;467:849–853.
Heinrich MC, Corless CL, Demetri GD et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–4349.
Antonescu CR, Besmer P, Guo T et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005;11:4182–4190.
Chen LL, Holden JA, Choi H et al. Evolution from heterozygous to homozygous KIT mutation in gastrointestinal stromal tumor correlates with the mechanism of mitotic nondisjunction and significant tumor progression. Mod Pathol 2008;21:826–836.
Heinrich MC, Corless CL, Duensing A et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710.
Hirota S, Ohashi A, Nishida T et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003;125:660–667.
Kang HJ, Nam SW, Kim H et al. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene 2005;24:1066–1074.
Matei D, Satpathy M, Cao L et al. The platelet-derived growth factor receptor alpha is destabilized by geldanamycins in cancer cells. J Biol Chem 2007;282:445–453.
Debiec-Rychter M, Wasag B, Stul M et al. Gastrointestinal stromal tumours (GISTs) negative for KIT (CD117 antigen) immunoreactivity. J Pathol 2004;202:430–438.
Duensing A, Joseph NE, Medeiros F et al. Protein kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res 2004;64:5127–5131.
Wozniak A, Sciot R, Guillou L et al. Array CGH analysis in primary gastrointestinal stromal tumors: cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer 2007;46:261–276.
Subramanian S, West RB, Corless CL et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene 2004;23:7780–7790.
Wasag B, Debiec-Rychter M, Pauwels P et al. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol 2004;17:889–894.
Medeiros F, Corless CL, Duensing A et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol 2004;28:889–894.
Sakurai S, Hasegawa T, Sakuma Y et al. Myxoid epithelioid gastrointestinal stromal tumor (GIST) with mast cell infiltrations: a subtype of GIST with mutations of platelet-derived growth factor receptor alpha gene. Hum Pathol 2004;35:1223–1230.
Wardelmann E, Hrychyk A, Merkelbach-Bruse S et al. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn 2004;6:197–204.
Lasota J, Dansonka-Mieszkowska A, Sobin LH et al. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest 2004;84:874–883.
Hostein I, Faur N, Primois C et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 2010;133:141–148.
Andersson J, Sihto H, Meis-Kindblom JM et al. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 2005;29:1170–1176.
de Raedt T, Cools J, Debiec-Rychter M et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from a dominant activating mutation in PDGFRA. Gastroenterology 2006;131:1907–1912.
Kang DY, Park CK, Choi JS et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007;31:224–232.
Maertens O, Prenen H, Debiec-Rychter M et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006;15:1015–1023.
Miettinen M, Fetsch JF, Sobin LH et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006;30:90–96.
Stewart C, Corless C, Rubin BP et al. Mitotic recombination as evidence of alternative pathogenesis of gastrointestinal stromal tumours in neurofibromatosis type 1. J Med Genet 2007;44:e61.
Kinoshita K, Hirota S, Isozaki K et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol 2004;202:80–85.
Miettinen M, Killian JK, Wang ZF et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol 2012;37:234–240.
Mason E, Hornick JL . Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol advance online publication, 7 June 2013 (e-pub ahead of print).
Killian JK, Kim SY, Miettinen M et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 2013;3:648–657.
Pantaleo MA, Astolfi A, Indio V et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011;103:983–987.
Wagner AJ, Remillard SP, Zhang YX et al. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol 2013;26:289–294.
Dwight T, Benn DE, Clarkson A et al. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol 2012;37:226–233.
Pantaleo MA, Astolfi A, Urbini M et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet 2013, (e-pub ahead of print).
Oudijk L, Gaal J, Korpershoek E et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol 2013;3:456–463.
Italiano A, Chen CL, Sung YS et al. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer 2012;14:408.
Belinsky MG, Rink L, Flieder DB et al. Overexpression of insulin-like growth factor 1 receptor and frequent mutational inactivation of SDHA in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer 2013;52:214–224.
Nannini M, Astolfi A, Paterini P et al. Expression of IGF-1 receptor in KIT/PDGF receptor-a wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol 2013;9:121–126.
Prakash S, Sarran L, Socci N et al. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol 2005;27:179–187.
Janeway KA, Liegl B, Harlow A et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res 2007;67:9084–9088.
Agaram NP, LaQuaglia MP, Ustun B et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res 2008;14:3204–3215.
Carney JA . Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 1999;74:543–552.
Beghini A, Tibiletti MG, Roversi G et al. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001;92:657–662.
Chompret A, Kannengiesser C, Barrois M et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004;126:318–321.
Hirota S, Nishida T, Isozaki K et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology 2002;122:1493–1499.
Isozaki K, Terris B, Belghiti J et al. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol 2000;157:1581–1585.
Maeyama H, Hidaka E, Ota H et al. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology 2001;120:210–215.
Nishida T, Hirota S, Taniguchi M et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998;19:323–324.
Agaimy A, Wunsch PH, Hofstaedter F et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol 2007;31:113–120.
Corless CL, McGreevey L, Haley A et al. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol 2002;160:1567–1572.
Kawanowa K, Sakuma Y, Sakurai S et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol 2006;37:1527–1535.
Muenst S, Thies S, Went P et al. Frequency, phenotype, and genotype of minute gastrointestinal stromal tumors in the stomach: an autopsy study. Hum Pathol 2011;42:1849–1854.
Rossi S, Gasparotto D, Toffolatti L et al. Molecular and clinicopathologic characterization of gastrointestinal stromal tumors (GISTs) of small size. Am J Surg Pathol 2010;34:1480–1491.
Gasparotto D, Rossi S, Bearzi I et al. Multiple primary sporadic gastrointestinal stromal tumors in the adult: an underestimated entity. Clin Cancer Res 2008;14:5715–5721.
Bergmann F, Gunawan B, Hermanns B et al. Cytogenetic and morphologic characteristics of gastrointestinal stromal tumors. Recurrent rearrangement of chromosome 1 and losses of chromosomes 14 and 22 as common anomalies. Verh Dtsch Ges Pathol 1998;82:275–278.
Debiec-Rychter M, Lasota J, Sarlomo-Rikala M et al. Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet 2001;128:24–30.
Fukasawa T, Chong JM, Sakurai S et al. Allelic loss of 14q and 22q, NF2 mutation, and genetic instability occur independently of c-kit mutation in gastrointestinal stromal tumor. Jpn J Cancer Res 2000;91:1241–1249.
Heinrich MC, Rubin BP, Longley BJ et al. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol 2002;33:484–495.
Kim NG, Kim JJ, Ahn JY et al. Putative chromosomal deletions on 9P, 9Q and 22Q occur preferentially in malignant gastrointestinal stromal tumors. Int J Cancer 2000;85:633–638.
Assamaki R, Sarlomo-Rikala M, Lopez-Guerrero JA et al. Array comparative genomic hybridization analysis of chromosomal imbalances and their target genes in gastrointestinal stromal tumors. Genes Chromosomes Cancer 2007;46:564–576.
Gunawan B, von HA, Sander B et al. An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J Pathol 2007;211:463–470.
El Rifai W, Sarlomo-Rikala M, Miettinen M et al. DNA copy number losses in chromosome 14: an early change in gastrointestinal stromal tumors. Cancer Res 1996;56:3230–3233.
O'Leary T, Ernst S, Przygodzki R et al. Loss of heterozygosity at 1p36 predicts poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1999;79:1461–1467.
Schurr P, Wolter S, Kaifi J et al. Microsatellite DNA alterations of gastrointestinal stromal tumors are predictive for outcome. Clin Cancer Res 2006;12:5151–5157.
el-Rifai W, Sarlomo-Rikala M, Andersson LC et al. High-resolution deletion mapping of chromosome 14 in stromal tumors of the gastrointestinal tract suggests two distinct tumor suppressor loci. Genes Chromosomes Cancer 2000;27:387–391.
Yamamoto H, Tobo T, Nakamori M et al. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol 2009;135:791–798.
Hur K, Lee HJ, Woo JH et al. Gene expression profiling of human gastrointestinal stromal tumors according to its malignant potential. Dig Dis Sci 2010;55:2561–2567.
Perrone F, Tamborini E, Dagrada GP et al. 9p21 locus analysis in high-risk gastrointestinal stromal tumors characterized for c-kit and platelet-derived growth factor receptor alpha gene alterations. Cancer 2005;104:159–169.
Ricci R, Arena V, Castri F et al. Role of p16/INK4a in gastrointestinal stromal tumor progression. Am J Clin Pathol 2004;122:35–43.
Sabah M, Cummins R, Leader M et al. Loss of heterozygosity of chromosome 9p and loss of p16INK4A expression are associated with malignant gastrointestinal stromal tumors. Mod Pathol 2004;17:1364–1371.
Schneider-Stock R, Boltze C, Lasota J et al. Loss of p16 protein defines high-risk patients with gastrointestinal stromal tumors: a tissue microarray study. Clin Cancer Res 2005;11 (2 Pt 1):638–645.
Feakins RM . The expression of p53 and bcl-2 in gastrointestinal stromal tumours is associated with anatomical site, and p53 expression is associated with grade and clinical outcome. Histopathology 2005;46:270–279.
Panizo-Santos A, Sola I, Vega F et al. Predicting metastatic risk of gastrointestinal stromal tumors: role of cell proliferation and cell cycle regulatory proteins. Int J Surg Pathol 2000;8:133–144.
Romeo S, Debiec-Rychter M, Van Glabbeke M et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res 2009;15:4198.
Tornillo L, Duchini G, Carafa V et al. Patterns of gene amplification in gastrointestinal stromal tumors (GIST). Lab Invest 2005;85:921–931.
Druker BJ, Tamura S, Buchdunger E et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561–566.
Joensuu H, Roberts PJ, Sarlomo-Rikala M et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001;344:1052–1056.
Van Oosterom AT, Judson I, Verweij J et al. STI571, an active drug in metastatic gastrointestinal stromal tumors (GIST) an EORTC phase I study [abstract]. Proc Am Soc Clin Oncol 2001;20:1a.
Demetri GD, von Mehren M, Blanke CD et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480.
Blanke CD, Rankin C, Demetri GD et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626–632.
Verweij J, Casali PG, Zalcberg J et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004;364:1127–1134.
Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol 2010;28:1247–1253.
von Mehren M, Heinrich MC, Joensuu H et al. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST). J Clin Oncol 2011;29:609s (Suppl) abstr 10016.
DeMatteo RP, Ballman KV, Antonescu CR et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:1097–1104.
Joensuu H, Eriksson M, Sundby Hall K et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA 2012;307:1265–1272.
Cassier PA, Ducimetière F, Lurkin A et al. A prospective epidemiological study of new incident GISTs during two consecutive years in Rhône Alpes region: incidence and molecular distribution of GIST in a European region. Br J Cancer 2010;103:165–170.
Miettinen M, Sobin LH, Lasota J . Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005;29:52–68.
Miettinen M, Makhlouf H, Sobin LH et al. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006;30:477–489.
Miettinen M, Kopczynski J, Makhlouf HR et al. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the duodenum: a clinicopathologic, immunohistochemical, and molecular genetic study of 167 cases. Am J Surg Pathol 2003;27:625–641.
Agaimy A, Vassos N, Märkl B et al. Anorectal gastrointestinal stromal tumors: a retrospective multicenter analysis of 15 cases emphasizing their high local recurrence rate and the need for standardized therapeutic approach. Int J Colorectal Dis 2013;28:1057–1064.
Le Cesne A, Ray-Coguard I, Bui BN et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol 2010;11:942–949.
Agaram NP, Besmer P, Wong GC et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res 2007;13:170–181.
Antonescu CR, Romeo S, Zhang L et al. Dedifferentiation in gastrointestinal stromal tumor to an anaplastic KIT-negative phenotype: a diagnostic pitfall: morphologic and molecular characterization of 8 cases occurring either de novo or after imatinib therapy. Am J Surg Pathol 2013;37:385–392.
Heinrich MC, Owzar K, Corless CL et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360–5367.
Debiec-Rychter M, Sciot R, Le CA et al. Kit mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093–1103.
Heinrich MC, Maki RG, Corless CL et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–5359.
Corless CL, Schroeder A, Griffith D et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357–5364.
Weisberg E, Wright RD, Jiang J et al. Effects of PKC412, nilotinib, and imatinib against GIST-associated PDGFRA mutants with differential imatinib sensitivity. Gastroenterology 2006;131:1734–1742.
Gajiwala KS, Wu JC, Christensen J et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci USA 2009;106:1542–1547.
Cassier PA, Fumagalli E, Rutkowski P et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res 2012;18:4458–4464.
Agaimy A, Terracciano LM, Dirnhofer S et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol 2009;62:613–616.
Agaram NP, Wong GC, Guo T et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008;47:853–859.
Janeway KA, Albritton KH, Van den Abbeele AD et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 2009;52:767–771.
Chen LL, Trent JC, Wu EF et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 2004;64:5913–5919.
Debiec-Rychter M, van OA, Marynen P . Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005;128:270–279.
Grimpen F, Yip D, McArthur G et al. Resistance to imatinib, low-grade FDG-avidity on PET, and acquired KIT exon 17 mutation in gastrointestinal stromal tumour. Lancet Oncol 2005;6:724–727.
Heinrich MC, Corless CL, Blanke CD et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006;24:4764–4774.
Koyama T, Nimura H, Kobayashi K et al. Recurrent gastrointestinal stromal tumor (GIST) of the stomach associated with a novel c-kit mutation after imatinib treatment. Gastric Cancer 2006;9:235–239.
Wakai T, Kanda T, Hirota S et al. Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumour is associated with a second KIT mutation. Br J Cancer 2004;90:2059–2061.
Wardelmann E, Thomas N, Merkelbach-Bruse S et al. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol 2005;6:249–251.
Liegl B, Kepten I, Le C et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64–74.
Nishida T, Kanda T, Nishitani A et al. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci 2008.
Lim KH, Huang MJ, Chen LT et al. Molecular analysis of secondary kinase mutations in imatinib-resistant gastrointestinal stromal tumors. Med Oncol 2008;25:207–213.
Demetri GD . Differential properties of current tyrosine kinase inhibitors in gastrointestinal stromal tumors. Semin Oncol 2011;38 (Suppl 1):S10–S19.
Demetri GD, Van Oosterom AT, Garrett CR et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006;368:1329–1338.
Nishida T, Takahashi T, Nishitani A et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol 2009;14:143–149.
Demetri GD, Reichardt P, Kang Y et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. [abstract]. Lancet 2013;381:295–302.
Eide CA, Adrian LT, Tyner JW et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res 2011;71:3189–3195.
Heinrich MC, Wise S, Hood M et al. In vitro activity of novel KIT/PDGFRA switch pocket kinase inhibitors against mutations associated with drug-resistant GI stromal tumors. J Clin Oncol 2010;28:15s (Suppl) abstr 10007.
Lasota J, Miettinen M . KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol 2006;23:91–102.
Acknowledgements
We thank all the members, past and present, of the Corless and Heinrich laboratories at the Oregon Health and Science University for their continuing efforts on GIST research. Some of the works referenced in this article were supported by generous donations from the GIST Cancer Research Fund and the BP Lester Foundation, and by grant support from the LifeRaft Group.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
CLC has received honoraria, consulting fees, and research support from Novartis and Pfizer. CLC has also received a consulting fee from Bayer.
Rights and permissions
About this article
This article is cited by
-
Personalized radiomics signature to screen for KIT-11 mutation genotypes among patients with gastrointestinal stromal tumors: a retrospective multicenter study
Journal of Translational Medicine (2023)
-
Metanephric stromal tumor in an adult with PDGFRA mutation: a case report
Diagnostic Pathology (2023)
-
Liver resection for metastatic GIST tumor improves survival in the era of tyrosine kinase inhibitors: a systematic review and meta-analysis
Langenbeck's Archives of Surgery (2023)
-
Effect of Verapamil, a P-glycoprotein-1 and Cytochrome P450 3A4 Inhibitor, on Pharmacokinetics and Metabolic Stability of Ripretinib: A Drug–Drug Interaction Study in Rats
European Journal of Drug Metabolism and Pharmacokinetics (2023)
-
Unique gastrointestinal stromal tumor with PDGFRA D842Y mutation—evaluation of in vivo sensitivity to imatinib
memo - Magazine of European Medical Oncology (2021)
