
Abstract
IgG4-related disease is a newly recognized fibro-inflammatory condition characterized by several features: a tendency to form tumefactive lesions in multiple sites; a characteristic histopathological appearance; and—often but not always—elevated serum IgG4 concentrations. An international symposium on IgG4-related disease was held in Boston, MA, on 4–7 October 2011. The organizing committee comprising 35 IgG4-related disease experts from Japan, Korea, Hong Kong, the United Kingdom, Germany, Italy, Holland, Canada, and the United States, including the clinicians, pathologists, radiologists, and basic scientists. This group represents broad subspecialty expertise in pathology, rheumatology, gastroenterology, allergy, immunology, nephrology, pulmonary medicine, oncology, ophthalmology, and surgery. The histopathology of IgG4-related disease was a specific focus of the international symposium. The primary purpose of this statement is to provide practicing pathologists with a set of guidelines for the diagnosis of IgG4-related disease. The diagnosis of IgG4-related disease rests on the combined presence of the characteristic histopathological appearance and increased numbers of IgG4+ plasma cells. The critical histopathological features are a dense lymphoplasmacytic infiltrate, a storiform pattern of fibrosis, and obliterative phlebitis. We propose a terminology scheme for the diagnosis of IgG4-related disease that is based primarily on the morphological appearance on biopsy. Tissue IgG4 counts and IgG4:IgG ratios are secondary in importance. The guidelines proposed in this statement do not supplant careful clinicopathological correlation and sound clinical judgment. As the spectrum of this disease continues to expand, we advocate the use of strict criteria for accepting newly proposed entities or sites as components of the IgG4-related disease spectrum.
Similar content being viewed by others

Introduction and historical perspective
IgG4-related disease is a newly recognized fibro-inflammatory condition characterized by several features: a tendency to form tumefactive lesions at multiple sites; a dense lymphoplasmacytic infiltrate rich in IgG4+ plasma cells; storiform fibrosis; and—often but not always—elevated serum IgG4 concentrations.1, 2 The disease was initially recognized in the pancreas, disease now known as autoimmune pancreatitis. Autoimmune pancreatitis was linked with the presence of elevated levels of serum IgG4 in 2001.1
Two distinct disorders within the category currently known as autoimmune pancreatitis are recognized. Type 1 autoimmune pancreatitis (now sometimes termed as ‘IgG4-related pancreatitis’) demonstrates the classic histological features of IgG4-related disease.3, 4 In contrast, type 2 autoimmune pancreatitis shows little similarity to IgG4-related disease. Type 2 autoimmune pancreatitis is associated with neutrophilic infiltrates and (occasionally) epithelioid cell granulomas, both of which are generally inconsistent with the diagnosis of IgG4-related disease, as described below.3
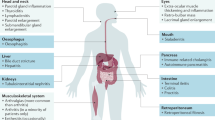
The observation that patients with autoimmune pancreatitis have extrapancreatic fibro-inflammatory lesions rich in IgG4-bearing cells, either synchronous or metachronous, with similar findings in other organs, led to the concept of IgG4-related disease.5 This condition has now been described in virtually every organ system: the biliary tree, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid, pericardium, and skin.2 The histopathological features bear striking similarities across the involved organs.6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 IgG4-related disease is therefore analogous to sarcoidosis, another systemic disease in which diverse organ manifestations are linked by a unique histological appearance. Disparate disorders such as Mikulicz's syndrome, Küttner's tumor, multifocal fibrosclerosis, and eosinophilic angiocentric fibrosis are now considered to fall within this disease spectrum.7, 17, 18, 19 Although their presenting symptoms are highly variable, the features that tie these disease conditions together are a common histological appearance, elevated serum and tissue levels of IgG4, and a response, generally swift, to immunosuppression.
The nomenclature of IgG4-related disease has continued to evolve. A group of Japanese investigators recently elected to use the term ‘IgG4-related disease’ for this condition, in preference to such potential alternatives as IgG4-related systemic disease, IgG4-related sclerosing disease, and IgG4-related multi-organ lymphoproliferative syndrome.20 We acknowledge that much remains unknown about the behavior of the IgG4 molecule in vivo, the pathways through which this immunoglobulin participates in disease, and whether or not the role of IgG4 is primary or secondary.20, 21, 22 In time, discoveries pertaining to the etiology and pathophysiology of this condition may lead to proposals for nomenclature that is deemed more appropriate. For the present, the term ‘IgG4-related disease’ aptly recognizes the ubiquity of IgG4 within involved organs and the frequency of elevated serum IgG4 concentrations. Until a more detailed understanding of this condition suggests a better name, we endorse the term IgG4-related disease.
An international symposium on IgG4-related disease was held in Boston, Massachusetts on 4–7 October 2011 (http://www2.massgeneral.org/pathology/symposium/IgG4_related_systemic_dis.asp). The organizing committee comprising 35 IgG4-related disease experts from Japan, South Korea, Hong Kong, the United Kingdom, Germany, Italy, Holland, Canada, and the United States, including the clinicians, pathologists, radiologists, and basic scientists. This group represents broad subspecialty expertise in pathology, rheumatology, gastroenterology, allergy, immunology, nephrology, pulmonary medicine, oncology, ophthalmology, and surgery. The members were chosen based on their contributions to the literature. The histopathology of IgG4-related disease was a specific focus of the international symposium. The primary purpose of this statement is to provide practicing pathologists with a set of guidelines for the diagnosis of IgG4-related disease. These guidelines are endorsed by the committee listed on this paper. We recognize that the guidelines will require revision as additional data and new biomarkers become available.
These guidelines are not intended to supplant proposals for organ-specific diagnostic criteria in IgG4-related disease.20, 23, 24, 25 The lesions histologically suggestive of IgG4-related disease based on these guidelines will often fulfill organ-specific criteria, in which histology is a key component. Discrepancies, if any, must be resolved in an interdisciplinary setting and with further evaluation, including a rebiopsy if necessary. We endorse the view that diagnostic schemes focused upon individual organs may improve accuracy for these disorders, because a certain variability in the pathological findings exists across the organs. We describe some specific examples of this below.
Although the combination of histopathological features and immunohistochemical stain results can provide strong supportive evidence for the diagnosis of IgG4-related disease, careful correlation with the clinical scenario and imaging characteristics of a particular patient is often required to arrive at a definitive diagnosis. Thus, when referring to conclusions that can be made from the interpretation of pathology results alone, we avoid terms such as ‘definite’, preferring instead ‘histologically suggestive of IgG4-related disease’.
Histopathological features of IgG4-related disease
The Centrality of Morphological Findings
The two features that link the disparate manifestations of IgG-related disease are a characteristic histopathological appearance and an elevated number of IgG4+ plasma cells within tissue. The serum IgG4 concentration is elevated in many patients—often dramatically—but serum concentrations of this immunoglobulin are normal in up to 40% of patients with biopsy-proven IgG4-related disease.26 Moreover, neither an increase in serum IgG4 nor the finding of elevated numbers of IgG4+ plasma cells in tissue is specific for IgG4-related disease. In the absence of a more specific biomarker, we support the position that in the appropriate clinical context, morphological features form the fundamental basis for the diagnosis of IgG4-related disease. However, the diagnosis of IgG4-related disease cannot be established with certainty in the absence of an immunohistochemical stain for IgG4. Thus, the diagnosis of IgG4-related disease requires both an appropriate histological appearance and increased numbers of IgG4+ plasma cells (or an elevated IgG4:IgG ratio) in tissue.
The three major histopathological features associated with IgG4-related disease are (Figure 1)3, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17

Characteristic histological features of IgG4-related disease. (a) IgG4-related sialadenitis. The salivary gland is extensively infiltrated by inflammatory cells, which consist of lymphocytes and plasma cells. (b) IgG4-related sialadenitis. A moderate number of eosinophils are present. H&E, × 400. (c) IgG4-related orbital disease. IgG4-related disease typically shows an irregularly whorled pattern of fibrosis (storiform fibrosis). (d) Type 1 autoimmune pancreatitis (IgG4-related pancreatitis). The vein (*) is completely obliterated by aggregated inflammatory cell infiltration (obliterative phlebitis). The adjacent artery is patent. However, the obliterated vein is not readily identified on an H&E stain and unequivocal evidence of an obliterated vein is only seen on the elastin stain (e), × 100. (f) Type 1 autoimmune pancreatitis (IgG4-related pancreatitis). The vein shows transmural infiltration by inflammatory cells, but is not associated with luminal obliteration.
-
1)
Dense lymphoplasmacytic infiltrate
-
2)
Fibrosis, arranged at least focally in a storiform pattern
-
3)
Obliterative phlebitis
Other histopathological features associated with IgG4-related disease are (Figure 1)
-
1)
Phlebitis without obliteration of the lumen
-
2)
Increased numbers of eosinophils
However, in isolation, these latter two features are neither sensitive nor specific for the diagnosis of IgG4-related disease.
In most instances, a confident pathological diagnosis of IgG4-related disease requires the presence of two of the three major histological features. In the majority of cases, these include a dense lymphoplasmacytic infiltrate and storiform-type fibrosis. Exceptions to this rule exist, however, in organs such as the lymph node,27 lung,9 minor salivary glands, and lacrimal glands.28 In those organs, storiform-type fibrosis or obliterative phlebitis may be inconspicuous or absent (see Table 1).
Histopathological Features of Key Findings
We provide some definitions of histopathological features that are relevant to the diagnosis of IgG4-related disease.3, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17
Dense lymphoplasmacytic infiltrate
The majority of cells are small lymphocytes that are distributed diffusely throughout the lesion and intermingled with plasma cells (Figure 1a and b). Germinal centers are observed occasionally. The lymphocytic infiltrate is composed predominantly of T cells, with scattered aggregates of B cells. Plasma cells are an essential component and may predominate. Eosinophils are found in mild to moderate quantities (Figure 1b) and dominate in a minority of cases, particularly in the setting of eosinophilic angiocentric fibrosis.7 Scattered macrophages may also be present.
Storiform-type fibrosis
The storiform-type pattern resembles the spokes of a cartwheel with spindle cells radiating from a center (Figure 1c). The spindle cells, which are either fibroblasts or myofibroblasts, are typically buried within the lymphoplasmacytic infiltrate. The storiform pattern of fibrosis may not be detected in limited samples such as needle biopsies.
Obliterative phlebitis
The venous channels are obliterated by a dense lymphoplasmacytic infiltrate (Figure 1d–e). Lymphocytes and plasma cells are seen both within the wall of the venous channel and within the lumen. Partially obliterated veins with transmural inflammatory infiltrates are also consistent with the diagnosis of IgG4-related disease (Figure 1f). Fully obliterated veins may require elastin stains for identification. However, medium-sized venous channels are generally accompanied by arteries (Figure 1e), which are less likely to be affected by the inflammatory process and can therefore serve as a guidepost to detecting obliterated venous structures. Obliterated venous channels without the requisite inflammation are not considered as evidence of IgG4-related disease.
The presence of arteritis does not exclude the diagnosis of IgG4-related disease. Arteritis is occasionally observed in cases of autoimmune pancreatitis and in the lung lesions of IgG4-related disease. The arteritis of IgG4-related disease is characterized by a non-necrotizing lymphoplasmacytic infiltrate with or without obliteration of the lumen, similarly to obliterative phlebitis. Necrotizing forms of arteritis are not seen.
Histopathological Features Inconsistent with a Diagnosis of IgG4-Related Disease
The two features that are relatively inconsistent with the diagnosis of IgG4-related disease are the presence of epithelioid cell granulomas and a prominent neutrophilic infiltrate.6 The presence of granulomas generally excludes the diagnosis of IgG4-related disease except when the granulomas represent a coexisting lesion/disease that occurs in a background typical for IgG4-related disease.2, 3, 6 Similarly, giant cells are identified only rarely in this disease. Neutrophilic microabscesses and zones of necrosis are also not central features of IgG4-related disease, except in the presence of erosion and ulceration, particularly in the upper aerodigestive tract. In the lung, small aggregates of neutrophils can be present in bronchioloalveolar spaces.9
The presence of neutrophils, necrosis, and giant cells raises the specter of granulomatosis with polyangiitis (formerly Wegener's).
Quantitative assessment of the IgG4 stain
IgG4 immunostaining is an essential test for the pathological diagnosis of IgG4-related disease (Figure 2a and b). This applies particularly to cases without an elevated concentration of serum IgG4. One may argue that immunostaining is not necessary for straightforward cases such as specimens obtained at the time of a Whipple procedure or IgG4-related sialadenitis. However, IgG4 immunostaining is strongly recommended even in those cases because it is a simple, highly reproducible test that provides strong confirmatory evidence for the diagnosis.

Immunostaining for IgG and IgG4 in IgG4-related dacryoadenitis. The majority of IgG-positive plasma cells (a) appear positive for IgG4 (b). (a) IgG immunostaining and (b) IgG4 immunostaining, both at × 200.
The Appropriate Cutoff for the Number of IgG4+ Plasma Cells
In IgG4-related pancreatitis (type 1 autoimmune pancreatitis), the finding of >30 IgG4+ plasma cells per high-power field (hpf) has been reported to have acceptable specificity.29, 30, 31, 32 Furthermore, dense, diffuse infiltrates of IgG4+ plasma cells that number >50/hpf are reportedly highly specific.6, 10, 29, 30, 31, 32 On biopsy specimens, the presence of >10 IgG4+ plasma cells has been proposed as one component of a comprehensive diagnostic panel.33 However, the appropriate cutoff point may vary from organ to organ because of the predominance of fibrosis at the time the diagnosis is made. A cardinal example of this is IgG4-related retroperitoneal fibrosis. Furthermore, the availability of published data at some sites is limited. We have proposed a set of cutoff points that is specific to each organ (Figure 3). It is worth re-emphasizing here that a sample attaining the threshold for the IgG4 immunoperoxidase stain does not necessarily qualify for the diagnosis of IgG4-related disease. The diagnosis of IgG4-related disease requires careful and deliberate correlation with the histopathological features in the sample, as well as with the clinical and radiological findings.

The IgG4-to-IgG Ratio
IgG4+/IgG+ plasma cell ratio is a more powerful tool than IgG4+ plasma cell counts in establishing the diagnosis of IgG4-related disease.6, 9, 11, 27, 34, 35, 36, 37 As noted, some inflammatory lesions that are not IgG4-related disease are associated with high numbers of IgG4+ plasma cells per hpf simply because of the abundance of plasma cells.11 Therefore, the IgG4+ plasma cell count alone may not help to distinguish between IgG4-related disease and disorders that are not part of that disease spectrum.
Some researchers have suggested an IgG4+/IgG+ plasma cell ratio of >40% as a comprehensive cutoff value in any organ.11, 27, 37 In fact, at most sites of documented IgG4-related disease, the IgG4+/IgG+ plasma cell ratio is >40%.6, 9, 11, 27, 34, 35, 36, 37 Moreover, in Japan, a consensus has been reached to adopt this as a histological diagnostic criterion for IgG4-related disease.25 However, in the absence of other corroborative findings, we do not accept an IgG4+/IgG+ plasma cell ratio of >40% in and of itself as sufficient pathological evidence of IgG4-related disease. This applies particularly to cases with a low overall IgG4 count per hpf. As an example, a case with 5 IgG4+ plasma cells/hpf and 10 IgG+ plasma cells/hpf would have an IgG4+/IgG+ plasma cell ratio of 50%, but the pathological diagnosis of IgG4-related disease is untenable in the absence of classic histopathological features and a compatible clinical picture.
Several additional caveats also apply to elevated IgG4 to IgG ratios, because a variety of non-IgG4-related disease entities can have IgG4+/IgG+ plasma cell ratios of >40%. For example, conditions sometimes associated with elevated serum interleukin-6 (IL-6) concentrations such as multicentric Castleman's disease, rheumatoid arthritis, and other immune-mediated conditions sometimes occur with abundant IgG4+ plasma cells within tissue (IgG4+/IgG+ plasma cell ratio >40%) and elevated serum IgG4 concentrations.38, 39
Methods for Semiquantitative Analysis of IgG4 Immunostain
There is no gold standard approach for counting IgG4+ plasma cells. Although the IgG immunostain often suffers from high background staining, it works well on paraffin-embedded tissue with easily identifiable, intense cytoplasmic positivity and provides an indispensible adjunct to the diagnosis of IgG4-related disease. Accurate IgG4+/IgG+ ratios are sometimes difficult to obtain because of the high background IgG stain.
Most published studies do not specify the precise method used to count IgG4+ plasma cells, but Shrestha et al10 recently reported a detailed description of their method. We recommend quantitative analyses of the IgG4 and IgG stains and discourage simple ‘eyeballing’ of the slides. When extremely high numbers of IgG4+ plasma cells are seen, a ‘gestalt’ approach may be adequate for diagnosis (Figure 2). Two general methods of IgG4 counting appear to be appropriate. First, IgG4+ and IgG+ cells can be counted using the printed photographs of the same microscopic field at × 40 objective lens. Second, direct counting can be performed under the microscope. Both methods are acceptable and provide reliable measures.
Because the IgG4+ cell distribution may be patchy, counting only areas of intense IgG4 focus (‘hot spots’) might be more representative. Random counting might result in underestimation of IgG4+ cells if there are many intervening microscopic fields without IgG4+ cells. Generally, however, IgG4-related disease in most sites tends to show diffusely increased IgG4+ cells, as opposed to focal aggregates of IgG4+ cells in other diseases.
The optimal reporting approach is to indicate the exact size of microscopic field used for IgG4 counting. However, it is difficult to implement such precision in the everyday practice of surgical pathology. We acknowledge this challenge, along with other inexactitudes associated with quantitation of this stain such as the specification of the number of fields counted. An alternative approach is to adjust IgG4+ cell counts required for the diagnosis based on the field diameter of eyepieces (Supplementary Table 1).
Although no single method of counting has been shown to be superior to all others, we recommend counting three × 40 fields with the highest number of IgG4+ plasma cells and calculating the average number of IgG4+ plasma cells within these fields. The same three fields should be counted for the purpose of calculating the IgG4-to-IgG ratio.
Technical Issues—IgG4 Immunostain
Most pathology laboratories use a mouse monoclonal antibody against human IgG4 (clone HP6025), which is designed to bind to the Fc portion of IgG4 molecules.7, 10, 11, 31, 40, 41 Polyclonal antibodies have been also used by some groups. The type of antibody does not have a significant impact on the count of IgG4+ plasma cells. However, the monoclonal antibody is preferable because plasma cells are more clearly stained. Antigen retrieval with proteinase or heat treatment is helpful to produce high-contrast signals. Background staining is sometimes strong, particularly in IgG4-related disease, probably due to permeating serum IgG4 or secreted proteins from infiltrating plasma cells.
Other Biomarkers for IgG4-Related Disease
IgG4 is currently the single most reliable biomarker applicable on tissue sections. Few studies of other potential tissue biomarkers have been conducted. FOXP3+ regulatory T cells are known to be increased in IgG4-related disease.42 One study showed that the detection of FOXP3+ lymphocytes in biopsy specimens from the ampulla of Vater is useful for the diagnosis of autoimmune pancreatitis.43 However, the diagnostic value of FOXP3 immunostaining is limited because its specificity is lower than that of IgG4 immunostaining. Given the Th2-dominant immune reaction in IgG4-related disease,42 immunostaining for Th2-type cytokines or chemokines is another potential tool, but reliable antibodies that work on paraffin-embedded sections are not commercially available.
The IgG4+/IgG+ plasma cell ratio on immunostaining is widely used to assess a preferential shift to IgG4 production in affected sites. We do not endorse the use of IgG1 since published data are limited.
Non-IgG4-Related Disease Cases with Elevated Numbers of IgG4-Positive Cells
It is now well documented that a large number of conditions that are outside the IgG4-related disease spectrum of disease can be associated with increased numbers of IgG4+ plasma cells in tissue.
Inflammatory conditions
Inflammatory disease conditions potentially associated with an increased number of IgG4+ plasma cells include oral inflammatory diseases, primary sclerosing cholangitis, anti-neutrophil cytoplasmic antibody-associated vasculitis, rheumatoid arthritis, inflammatory bowel disease, rhinosinusitis, Rosai–Dorfman disease, splenic sclerosing angiomatoid nodular transformation, cutaneous plasmacytosis, perforating collagenosis, and autoimmune atrophic gastritis (pernicious anemia).39, 44, 45, 46, 47, 48 Many IgG4+ plasma cells can also be present in lymphoproliferative disorders such as multicentric Castleman's disease, or even in non-specific settings such as pulmonary abscess or biliary xanthogranulomatous inflammation.38, 49 However, none of these conditions consistently shows IgG4-rich inflammation and all lack the characteristic histopathological features of IgG4-related disease. These conditions fall outside the bounds of IgG4-related disease, despite the presence in some cases of increased numbers of IgG4+ plasma cells.
Lymphoma
Low-grade B-cell lymphomas must be excluded in cases of possible IgG4-related disease with florid lymphoplasmacytic infiltrates, especially if the plasma cells exhibit atypical features such as prominent nuclear or cytoplasmic inclusions. The lymphomas that mimic IgG4-related disease are usually extranodal marginal zone lymphomas and sometimes follicular lymphomas and angioimmunoblastic lymphomas. The findings of sheets of CD20+ B cells or immunoglobulin light chain restriction support a diagnosis of lymphoma. As noted, the majority of lymphocytes in IgG4-related disease are T lymphocytes; B lymphocytes are usually found in nodular aggregates, often within reactive germinal centers.
Malignancies
Cancer tissue can be infiltrated by IgG4+ plasma cells to various degrees. Although published data are limited largely to pancreatobiliary cancers, this phenomenon may be seen in other malignancies as well.31, 32 IgG4+ plasma cell infiltration in malignant tissue is usually patchy, and not associated with other typical histological features of IgG4-related disease (eg, storiform fibrosis or obliterative phlebitis). IgG4+ plasma cells can also be identified in regional lymph nodes in cancer, although the exact frequency and nature of this phenomenon are still uncertain. The question of whether cases reported as synchronous carcinoma and IgG4-related disease represent a true association or non-specific peri-cancerous IgG4 reaction remains unresolved.50
The presence of elevated numbers of IgG4+ plasma cells within a tumor does not constitute sufficient evidence that the tumor arose in the setting of IgG4-related disease. Sometimes peritumoral tissue rich in IgG4+ plasma cells can mimic IgG4-related disease. Thus, a needle biopsy sampling the periphery of a malignant neoplasm may be misdiagnosed as IgG4-related disease. Careful examination of the available tissue for the histopathological features characteristic of IgG4-related disease helps avoid this diagnostic pitfall, but in equivocal cases it is usually appropriate to obtain more tissue.
Proposed diagnostic terminology for IgG4-related disease
We endorse a 3-tiered diagnostic terminology for the pathological diagnosis of IgG4-related disease (Figure 3). The underlying premise is that the histological features (dense lymphoplasmacytic infiltrate, fibrosis, arranged at least focally in a storiform pattern and obliterative phlebitis) associated with IgG4-related disease are highly specific when viewed in conjunction with an IgG4 stain. Nevertheless, correlation with the overall clinical scenario is required before an unequivocal diagnosis of IgG4-related disease can be established.
-
1)
Histologically highly suggestive of IgG4-related disease.
-
2)
Probable histological features of IgG4-related disease.
-
3)
Insufficient histopathological evidence of IgG4-related disease.
The histological features of IgG4-related disease are broadly similar across the various organs and organ systems but some sites (noted above) vary from this central paradigm. These criteria may not apply to certain sites of involvement, including lymph nodes, some examples of pulmonary IgG4-related disease, and biopsies from the oral mucosa (Table 1). The histological changes in these organs are varied and can mimic other diseases that are prevalent in these organs. Thus, the histopathological diagnosis at these sites relies considerably on the number of IgG4+ cells and on the ratio of IgG4 to IgG+ plasma cells.
Histologically Highly Suggestive of IgG4-Related Disease
This category requires the presence of at least two of the characteristic histological features listed below. One exception is the lacrimal gland, where both storiform fibrosis and obliterative phlebitis may be absent. Thus, one histological feature compatible with IgG4-related disease might suffice for dacryoadenitis.
-
1)
Dense lymphoplasmacytic infiltrate.
-
2)
Fibrosis, usually storiform in character.
-
3)
Obliterative phlebitis.
The IgG4 counts required for the diagnosis differ among affected organs, ranging from 10 to 200 cells/hpf. Surgical specimens generally show larger numbers of IgG4+ plasma cells than do needle biopsy specimens. An elevated IgG4+/IgG+ cell ratio of >40% is also necessary. When evaluating aortic specimens, we propose that a cell ratio of >50% be considered as a minimum criterion, because some cases of atherosclerosis and giant cell or infectious aortitis can show IgG4+/IgG+ ratios close to 40%.51, 52
The majority of cases that fulfill these criteria will show clinical and serological findings that are typical for IgG4-related disease. In the minority of cases that lack these criteria, histopathology would be the defining feature.
Probable Histological Features of IgG4-Related Disease
These cases either lack the full histological spectrum associated with IgG4-related disease or the immunohistochemical profile of IgG4-related disease. This category is also applied to organs where the concept of IgG4-related disease is not completely established. These cases generally fit into one of three clinical scenarios:
-
Cases with only a single histopathological feature, typically a dense lymphoplasmacytic infiltrate, and required numbers of IgG4+ cells. Some non-IgG4-related diseases fulfill these criteria.
-
Needle biopsies: Although needle biopsies often provide sufficient proof for the diagnosis of IgG4-RD, in some cases the complete histological picture is not represented on the biopsy sample. There is insufficient evidence to support the use of fine needle aspiration biopsy and cell block preparations for the diagnosis of IgG4-related disease.53
-
Meningeal and cutaneous disease: Published data for IgG4-related disease in these organs are limited.
Patients with diagnoses of histologically probable IgG4-related disease require additional clinical, serological, or radiological evidence to confirm the diagnosis of IgG4-related disease. Such additional evidence might include but is not limited to:
-
1)
Serum IgG4 >135 mg/dl.
-
2)
Other organ involvement, as demonstrated by radiological or pathological examination.
Insufficient Histopathological Evidence of IgG4-Related Disease
These cases are outside the two categories described above. Placement in this category does not necessarily exclude the diagnosis of IgG4-related disease entirely. Potential reasons include sampling artifact, the effects of previous therapy, and progression to a fibrotic stage.
Minimal Criteria to Propose IgG4-Related Disease Involvement of a New Organ/Site
To consider a previously unrecognized organ or site as being involved by IgG4-related disease, we recommend that the following criteria must be fulfilled: (1) characteristic histopathological findings with an elevated IgG4+ plasma cells and IgG4-to-IgG ratio; (2) high serum IgG4 concentrations; (3) effective response to glucocorticoid therapy; and (4) reports of other organ involvement that is consistent with IgG4-related disease.46, 54 Appropriate histopathological findings are essential, but they are not sufficient to establish a new manifestation/site of IgG4-related disease. In the early days of studies on IgG4-related disease, appropriate histopathological findings with only one additional criterion sufficed, usually elevated serum IgG4 or involvement of other organs.34, 35, 40
Conclusion
IgG4-related disease is a recently recognized multi-organ system condition with pathological features that are largely consistent across a wide range of organ systems. Although the precise role of IgG4 in this disease is unknown, its presence in tissue in association with plasma cells provides a robust biomarker for diagnosis when interpreted in the proper histopathological and clinical contexts.
The diagnosis of IgG4-related disease requires collaboration between the pathologist and the treating physician. This dialogue is critical in excluding the variety of other diseases that may show elevated serum and tissue levels of IgG4. The isolated presence of IgG4+ plasma cells or an elevated IgG4-to-IgG ratio constitutes relatively non-specific findings. The diagnosis of IgG4-related disease rests on the combined presence of the characteristic histopathological appearance and increased numbers of IgG4+ plasma cells. We propose a terminology scheme for the diagnosis of IgG4-related disease that is based primarily on the morphological appearance on biopsy. Tissue IgG4 counts and IgG4:IgG ratios are secondary in importance. The guidelines proposed in this statement do not supplant careful clinicopathological correlation and sound clinical judgment. As the spectrum of this disease continues to expand, we advocate use of strict criteria for accepting newly proposed entities or sites as components of the IgG4-related disease spectrum.
References
Hamano H, Kawa S, Horiuchi A, et al High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–738.
Stone JH, Zen Y, Deshpande V . IgG4-related disease. N Engl J Med 2012;366:539–551.
Deshpande V, Gupta R, Sainani NI, et al Subclassification of Autoimmune pancreatitis: a histologic classification with clinical significance. Am J Surg Pathol 2011;35:26–35.
Zamboni G, Luttges J, Capelli P, et al Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004;445:552–563.
Kamisawa T, Egawa N, Nakajima H . Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol 2003;98:2811–2812.
Zen Y, Nakanuma Y . IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol 2010;34:1812–1819.
Deshpande V, Khosroshahi A, Nielsen GP, et al Eosinophilic angiocentric fibrosis is a form of IgG4-related systemic disease. Am J Surg Pathol 2011;35:701–706.
Zen Y, Onodera M, Inoue D, et al Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol 2009;33:1833–1839.
Zen Y, Inoue D, Kitao A, et al IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2009;33:1886–1893.
Shrestha B, Sekiguchi H, Colby TV, et al Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with autoimmune pancreatitis: report of 6 and 12 cases with similar histopathology. Am J Surg Pathol 2009;33:1450–1462.
Sato Y, Kojima M, Takata K, et al Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman's disease. Mod Pathol 2009;22:589–599.
Deshpande V, Sainani NI, Chung RT, et al IgG4-associated cholangitis: a comparative histological and immunophenotypic study with primary sclerosing cholangitis on liver biopsy material. Mod Pathol 2009;22:1287–1295.
Chan SK, Cheuk W, Chan KT, et al IgG4-related sclerosing pachymeningitis: a previously unrecognized form of central nervous system involvement in IgG4-related sclerosing disease. Am J Surg Pathol 2009;33:1249–1252.
Cheuk W, Yuen HK, Chan AC, et al Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32:1159–1167.
Cornell LD, Chicano SL, Deshpande V, et al Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol 2007;31:1586–1597.
Kasashima S, Zen Y, Kawashima A, et al Inflammatory abdominal aortic aneurysm: close relationship to IgG4-related periaortitis. Am J Surg Pathol 2008;32:197–204.
Geyer JT, Ferry JA, Harris NL, et al Chronic sclerosing sialadenitis (Kuttner tumor) is an IgG4-associated disease. Am J Surg Pathol 2010;34:202–210.
Khosroshahi A, Bloch DB, Deshpande V, et al Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 2010;62:1755–1762.
Dahlgren M, Khosroshahi A, Nielsen GP, et al Riedel's thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken) 2010;62:1312–1318.
Umehara H, Okazaki K, Masaki Y, et al A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol 2012;22:1–14.
Nirula A, Glaser SM, Kalled SL, et al What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 2011;23:119–124.
Aalberse RC, Stapel SO, Schuurman J, et al Immunoglobulin G4: an odd antibody. Clin Exp Allergy 2009;39:469–477.
Raissian Y, Nasr SH, Larsen CP, et al Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011;22:1343–1352.
Kawano M, Saeki T, Nakashima H, et al Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol 2011;15:615–626.
Umehara H, Okazaki K, Masaki Y, et al Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol 2012;22:21–30.
Sah RP, Chari ST . Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol 2011;23:108–113.
Cheuk W, Yuen HK, Chu SY et al Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32:671–681.
Cheuk W, Yuen HK, Chan JK . Chronic sclerosing dacryoadenitis: part of the spectrum of IgG4-related Sclerosing disease? Am J Surg Pathol 2007;31:643–645.
Kamisawa T, Funata N, Hayashi Y, et al A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003;38:982–984.
Dhall D, Suriawinata AA, Tang LH, et al Use of immunohistochemistry for IgG4 in the distinction of autoimmune pancreatitis from peritumoral pancreatitis. Hum Pathol 2010;41:643–652.
Zhang L, Notohara K, Levy MJ, et al IgG4-positive plasma cell infiltration in the diagnosis of autoimmune pancreatitis. Mod Pathol 2007;20:23–28.
Deshpande V, Chicano S, Finkelberg D, et al Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol 2006;30:1537–1545.
Chari ST . Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing the Mayo Clinic's HISORt criteria. J Gastroenterol 2007;42 (Suppl 18):39–41.
Kitagawa S, Zen Y, Harada K, et al Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Kuttner's tumor). Am J Surg Pathol 2005;29:783–791.
Zen Y, Kitagawa S, Minato H, et al IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol 2005;36:710–717.
Sato Y, Notohara K, Kojima M, et al IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int 2010;60:247–258.
Cheuk W, Chan JK . IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303–332.
Sato Y, Kojima M, Takata K, et al Multicentric Castleman's disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol 2010;63:1084–1089.
Strehl JD, Hartmann A, Agaimy A . Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol 2011;64:237–243.
Zen Y, Harada K, Sasaki M, et al IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis-associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol 2004;28:1193–1203.
Cheuk W, Tam FK, Chan AN, et al Idiopathic cervical fibrosis—a new member of IgG4-related sclerosing diseases: report of 4 cases, 1 complicated by composite lymphoma. Am J Surg Pathol 2010;34:1678–1685.
Zen Y, Fujii T, Harada K, et al Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology 2007;45:1538–1546.
Kubota K, Kato S, Watanabe S, et al Usefulness of endoscopic biopsy using FOXP3(+) Treg up-regulation in the duodenal papilla in the differential diagnosis between autoimmune pancreatitis and pancreatic cancer. J Hepatobiliary Pancreat Sci 2011;18:414–421.
Narula N, Vasudev M, Marshall JK . IgG-related sclerosing disease: a novel mimic of inflammatory bowel disease. Dig Dis Sci 2010;55:3047–3051.
Moteki H, Yasuo M, Hamano H, et al IgG4-related chronic rhinosinusitis: a new clinical entity of nasal disease. Acta Otolaryngol 2011;131:518–526.
Zhang L, Smyrk TC . Autoimmune pancreatitis and IgG4-related systemic diseases. Int J Clin Exp Pathol 2010;3:491–504.
Kuo TT, Chen TC, Lee LY, et al IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease: an additional immunohistochemical feature and possible relationship to IgG4-related sclerosing disease. J Cutan Pathol 2009;36:1069–1073.
Kuo TT, Chen TC, Lee LY . Sclerosing angiomatoid nodular transformation of the spleen (SANT): clinicopathological study of 10 cases with or without abdominal disseminated calcifying fibrous tumors, and the presence of a significant number of IgG4+ plasma cells. Pathol Int 2009;59:844–850.
Zen Y, Fujii T, Sato Y, et al Pathological classification of hepatic inflammatory pseudotumor with respect to IgG4-related disease. Mod Pathol 2007;20:884–894.
Witkiewicz AK, Kennedy EP, Kennyon L, et al Synchronous autoimmune pancreatitis and infiltrating pancreatic ductal adenocarcinoma: case report and review of the literature. Hum Pathol 2008;39:1548–1551.
Stone JR . Aortitis, periaortitis, and retroperitoneal fibrosis, as manifestations of IgG4-related systemic disease. Curr Opin Rheumatol 2011;23:88–94.
Stone JH, Khosroshahi A, Deshpande V, et al IgG4-related systemic disease accounts for a significant proportion of thoracic lymphoplasmacytic aortitis cases. Arthritis Care Res (Hoboken) 2010;62:316–322.
Deshpande V, Mino-Kenudson M, Brugge WR, et al Endoscopic ultrasound guided fine needle aspiration biopsy of autoimmune pancreatitis: diagnostic criteria and pitfalls. Am J Surg Pathol 2005;29:1464–1471.
Kawa S, Okazaki K, Kamisawa T, et al Japanese consensus guidelines for management of autoimmune pancreatitis: II. Extrapancreatic lesions, differential diagnosis. J Gastroenterol 2010;45:355–369.
Lindstrom KM, Cousar JB, Lopes MB . IgG4-related meningeal disease: clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol 2010;120:765–776.
Detlefsen S, Mohr Drewes A, Vyberg M, et al. Diagnosis of autoimmune pancreatitis by core needle biopsy: application of six microscopic criteria. Virchows Arch 2009;454:531–539.
Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 2006;4:1010–1016; quiz 934.
Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology 2008;134:706–715.
Kawakami H, Zen Y, Kuwatani M, et al. IgG4-related sclerosing cholangitis and autoimmune pancreatitis: histological assessment of biopsies from Vater's ampulla and the bile duct. J Gastroenterol Hepatol 2010;25:1648–1655.
Umemura T, Zen Y, Hamano H, et al. Immunoglobin G4-hepatopathy: association of immunoglobin G4-bearing plasma cells in liver with autoimmune pancreatitis. Hepatology 2007;46:463–471.
Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 2010;78:1016–1023.
Cheuk W, Lee KC, Chong LY, et al. IgG4-related Sclerosing disease: a potential new etiology of cutaneous pseudolymphoma. Am J Surg Pathol 2009;33:1713–1719.
Khosroshahi A, Carruthers MD, Deshpande V, et al. Cutaneous immunoglobulin g4-related systemic disease. Am J Med 2011;124:e7–e8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article
Cite this article
Deshpande, V., Zen, Y., Chan, J. et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25, 1181–1192 (2012). https://doi.org/10.1038/modpathol.2012.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.72
Keywords
This article is cited by
-
IgG4-related disease: an update on pathology and diagnostic criteria with a focus on salivary gland manifestations
Virchows Archiv (2024)
-
Myofibroblast and pro-fibrotic cytokines in fibrosis of IgG4-related disease (IgG4-RD) patients from South Asia: preliminary data
Clinical Rheumatology (2024)
-
A case of IgG4-related interstitial nephritis with ureteral obstruction: case report and literature review
BMC Urology (2023)
-
A case report of hepatic angiomyolipoma, inflammatory subtype. Clinico-pathological characterization: a diagnostic challenge
Diagnostic Pathology (2023)
-
IgG4-related eosinophilic pleural effusion: a case report
BMC Geriatrics (2023)
