
Abstract
Approximately 45% of sporadic well-differentiated pancreatic neuroendocrine tumors harbor mutations in either ATRX (alpha thalassemia/mental retardation X-linked) or DAXX (death domain-associated protein). These novel tumor suppressor genes encode nuclear proteins that interact with one another and function in chromatin remodeling at telomeric and peri-centromeric regions. Mutations in these genes are associated with loss of their protein expression and correlate with the alternative lengthening of telomeres phenotype. Patients with multiple endocrine neoplasia-1 (MEN-1) syndrome, genetically defined by a germ line mutation in the MEN1 gene, are predisposed to developing pancreatic neuroendocrine tumors and thus represent a unique model for studying the timing of ATRX and DAXX inactivation in pancreatic neuroendocrine tumor development. We characterized ATRX and DAXX protein expression by immunohistochemistry and telomere status by telomere-specific fluorescence in situ hybridization in 109 well-differentiated pancreatic neuroendocrine lesions from 28 MEN-1 syndrome patients. The study consisted of 47 neuroendocrine microadenomas (<0.5 cm), 50 pancreatic neuroendocrine tumors (≥0.5 cm), and 12 pancreatic neuroendocrine tumor lymph node metastases. Expression of ATRX and DAXX was intact in all 47 microadenomas, and none showed the alternative lengthening of telomeres phenotype. ATRX and/or DAXX expression was lost in 3 of 50 (6%) pancreatic neuroendocrine tumors. In all three of these, tumor size was ≥3 cm, and loss of ATRX and/or DAXX expression correlated with the alternative lengthening of telomeres phenotype. Concurrent lymph node metastases were present for two of the three tumors, and each metastasis displayed the same changes as the primary tumor. These findings establish the existence of ATRX and DAXX defects and the alternative lengthening of telomeres phenotype in pancreatic neuroendocrine tumors in the context of MEN-1 syndrome. The observation that ATRX and DAXX defects and the alternative lengthening of telomeres phenotype occurred only in pancreatic neuroendocrine tumors measuring ≥3 cm and their lymph node metastases suggests that these changes are late events in pancreatic neuroendocrine tumor development.
Similar content being viewed by others


Main
Pancreatic neuroendocrine tumors are the second most common primary malignancy of the pancreas. Despite their improved prognosis as compared with pancreatic ductal adenocarcinoma, the survival rate is still only 65% at 5 years and 45% at 10 years.1
Although relatively little is known about the genetic development of pancreatic neuroendocrine tumors, recent studies have revealed that 43% of sporadic well-differentiated pancreatic neuroendocrine tumors harbor mutations in either ATRX (alpha thalassemia/mental retardation X-linked) or DAXX (death domain-associated protein).2 These two novel tumor suppressor genes encode nuclear proteins that interact with one another and are thought to function in chromatin remodeling at telomeric and peri-centromeric regions. Mutations in these genes are tightly associated with loss of nuclear expression of their respective proteins by immunohistochemistry and correlate with the alternative lengthening of telomeres phenotype, a telomerase-independent telomere maintenance mechanism.3, 4
Patients with multiple endocrine neoplasia-1 (MEN-1) syndrome have a germ line mutation in the MEN1 gene, which predisposes them to the development of pancreatic neuroendocrine tumors. The pancreata of these patients usually harbor multiple incidental neuroendocrine microadenomas (by definition measuring <0.5 cm), which are thought to represent precursors to pancreatic neuroendocrine tumors.5, 6 The MEN1 gene has also been shown to be somatically mutated in 44% of sporadic pancreatic neuroendocrine tumors,2 and up to 70% of sporadic pancreatic neuroendocrine tumors show chromosomal losses at 11q13, the MEN1 locus.7, 8, 9, 10, 11, 12, 13, 14, 15 Thus, on both histological and genetic levels, MEN-1 syndrome tumors are a rational model for studying the timing of genetic alterations in pancreatic neuroendocrine tumor development. For this reason, we characterized ATRX and DAXX protein expression (as a surrogate for gene status) and telomere status in 109 MEN-1 syndrome well-differentiated pancreatic neuroendocrine lesions.
Materials and methods
Design
This study was approved by the Internal Review Boards of The Johns Hopkins Hospital and the University Medical Center Utrecht. Twenty-eight patients with MEN-1 syndrome, diagnosed either by clinical history or by genetic testing, were identified through review of pathology files. Twenty patients were treated at The Johns Hopkins Hospital, and eight patients were treated at the University Medical Center Utrecht. Formalin-fixed paraffin-embedded tissue was available for all 28 patients. From these patients, 134 pancreatic neuroendocrine lesions were selected for characterization of ATRX and DAXX protein expression and telomere status. We sampled 1-11 lesions per patient (average of 4). Of these, 109 tumors, comprising 47 microadenomas, 50 pancreatic neuroendocrine tumors, and 12 pancreatic neuroendocrine tumor lymph node metastases had interpretable ATRX and DAXX immunolabeling results and telomere-specific fluorescence in situ hybridization (FISH) data. We adhered to the WHO 2010 Classification of the Tumors of the Gastrointestinal Tract nomenclature, which defines microadenomas as nonfunctional well-differentiated neuroendocrine neoplasms measuring <0.5 cm and pancreatic neuroendocrine tumors as neoplasms measuring ≥0.5 cm and/or functional tumors of any size.16 We had no functional tumors measuring less than 0.5 cm in our study. The hematoxylin and eosin and immunostained sections were reviewed by four pathologists (AM, GJAO, RHH, and KEM). Interpretation of the FISH data was performed independently by two other investigators (CMH and AKM). Immunolabeling and FISH data were interpreted independently (ie, without prior knowledge of the other data set).
Immunohistochemistry
Immunolabeling for ATRX and DAXX was performed as previously described,2 using the following antibodies and concentrations: anti-ATRX (HPA001906, Sigma-Aldrich, St Louis, MO, USA, 1:600); anti-DAXX (HPA008736, Sigma-Aldrich, 1:75 or 1:100). Briefly, formalin-fixed paraffin-embedded tumor sections were steamed with citrate buffer (pH 6.0) (Vector Laboratories, Burlingame, CA, USA) for 30 min to achieve antigen retrieval and then cooled for 10 min. Tissues were blocked against endogenous peroxidase activity with dual endogenous enzyme-blocking agent (Dako, Carpinteria, CA, USA) for 10 min. Sections were incubated with primary antibodies for 1 h at room temperature followed by secondary antibody (Leica Microsystems) for 30 min and detected with 3,3′-diaminobenzidine (Sigma-Aldrich) at 10 min. Wash steps were performed with phosphate buffered saline containing 0.1% Tween-20. Sections were counterstained with Harris' hematoxylin, rehydrated, and mounted. Immunolabeling for KI67 was performed as per standard laboratory protocol using the prediluted anti-KI67 antibody (clone 30-9, Ventana, Tucson, AZ, USA).
Immunolabeling for ATRX and DAXX was considered positive (ie, normal, intact) if at least 5% of neoplastic cells had nuclear labeling (and there was no evidence of cytoplasmic sequestration in the remaining cells). Neoplasms were scored as negative (ie, loss) for ATRX or DAXX if the pattern was that of cytoplasmic accumulation with nuclear clearing, as long as adequate internal controls (ie, nuclear labeling of adjacent endothelial cells, lymphocytes, and/or islets of Langerhans) were present. The designation ‘heterogeneous’ was applied to tumors that showed cytoplasmic sequestration of ATRX or DAXX in one subset of neoplastic cells and nuclear accumulation (>5%) in a different subset (regardless of whether the two subsets had similar or different morphology).
Fluorescence In Situ Hybridization
Formalin-fixed paraffin-embedded tumor sections were assayed for the alternative lengthening of telomeres phenotype by telomere-specific FISH as described previously.3, 17 Briefly, sections were de-paraffinized, hydrated, steamed in citrate buffer (Vector Laboratories) for 25 min, dehydrated, and then hybridized with a Cy3-labeled peptide nucleic acid probe complementary to the mammalian telomere repeat (CCCTAACCCTAACCCTAA). An Alexa Fluor 610-labeled peptide nucleic acid probe with specificity for the human centromeric CENP-B binding sequence (ATTCGTTGGAAACGGGA) was included as a positive control for hybridization. Sections were counterstained with DAPI to visualize nuclei.
Slides were analyzed with a Nikon 50i epifluorescence microscope with Xcite series 120 illuminator (EXFO Photonics Solutions) and appropriate excitation/emission filters, as previously described.17 Grayscale images were captured with Nikon NIS-Elements software 2.30 and a Photometrics Cool SNAP EZ digital camera, pseudo-colored, and merged. Tumors were considered positive for the alternative lengthening of telomeres phenotype if: (a) neoplastic cells demonstrated individual telomeric foci with integrated total signal intensities >10-fold that of the per cell mean integrated signal intensities for all telomeric signals in individual non-neoplastic stromal or endothelial cells within the same section, and (b) ≥1% of tumor cells displayed these alternative lengthening of telomeres-associated telomeric foci.17 Neoplasms were considered negative if they lacked the alternative lengthening of telomeres-associated telomeric foci and at least 500 neoplastic cells were analyzed. We did not assess co-localization of telomeric foci with promyelocytic leukemia nuclear bodies as this is not a requirement for the demonstration of the alternative lengthening of telomeres phenotype.3, 18, 19, 20
Statistical Analysis
A Fisher's exact test was used to determine if the difference in tumor grade between the ATRX or DAXX defective tumors and the tumors with intact ATRX and DAXX expression was statistically significant (P<0.05).
Results
Patient Demographics
The 28 MEN-1 syndrome patients consisted of 12 females and 16 males. The average age at the time of surgery was 45 years (range 19–81 years). All patients were Caucasian except one. Three patients underwent enucleation, 16 patients underwent distal pancreatectomy, and 9 patients underwent pancreaticoduodenectomy as their primary surgical intervention.
Pathological Findings
The mean size of the microadenomas was 0.2 cm. (range 0.1 to <0.5 cm); the mean size of the pancreatic neuroendocrine tumors was 1.9 cm (range 0.5–8.0 cm). Microscopically, the tumors showed a range of architectural patterns typical of neuroendocrine tumors, including trabecular, glandular, and solid patterns. The results of the ATRX, DAXX, and KI67 immunolabeling, the mitotic count, and the telomere-specific FISH are summarized in Table 1. Mitotic figures ranged from 0 to 5 per 10 high power fields with the vast majority of tumors (93%, 101/109) demonstrating fewer than two mitotic figures per 10 high power fields (WHO grade 1).21 KI67 expression ranged from 0 to 10% and showed a predominance of tumors (84%, 87/103) with a proliferative index ≤2% (WHO grade 1).21
Overall, 3% of the neuroendocrine lesions (microadenomas and pancreatic neuroendocrine tumors) showed defective ATRX and/or DAXX expression and displayed the alternative lengthening of telomeres phenotype. Of the 47 microadenomas, none had evidence of ATRX or DAXX defects or the alternative lengthening of telomeres phenotype (Figure 1a-d). Within the pancreatic neuroendocrine tumor group (ie, tumors measuring ≥0.5 cm), 3 of 50 tumors (6%) demonstrated defective ATRX and/or DAXX expression. Each of the three tumors was resected from a different MEN-1 syndrome patient. One tumor showed loss of DAXX expression (Figure 1e-g), a second tumor with prominent biphasic morphology demonstrated loss of nuclear DAXX in one morphological component and punctate nuclear immunolabeling for DAXX in the other morphological component (ie, heterogeneous staining pattern) (Figure 2a and b), and a third tumor showed a heterogeneous pattern of ATRX labeling and also loss of nuclear DAXX. Loss of nuclear expression of ATRX and/or DAXX correlated with the presence of the alternative lengthening of telomeres phenotype in all the three tumors. In addition, all three of the tumors with loss of ATRX and/or DAXX and the presence of the alternative lengthening of telomeres phenotype measured ≥3 cm. Two of the three tumors had concurrent lymph node metastases, and each metastasis showed the same changes as the primary tumor. No alterations in ATRX or DAXX expression or telomere status were identified in the remaining 10 lymph node metastases, which were associated with other pancreatic neuroendocrine tumors.

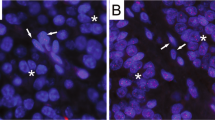
ATRX and DAXX immunolabeling and telomere-specific FISH in a MEN-1 microadenoma (a–d) and in a 3.5-cm MEN-1 pancreatic neuroendocrine tumor with a DAXX defect and the alternative lengthening of the telomeres phenotype (e–h). H&E (a, e), ATRX (b, f), DAXX with retained nuclear expression (c) and loss of nuclear expression (wih entrapped non-neoplastic duct) (g), telomere-specific FISH (d, h). Asterisks highlight nuclei with normal telomere-specific FISH signals. Arrows highlight alternative lengthening of telomeres-specific foci of telomeric DNA (DAPI nuclear counterstain). All images were taken at × 400 magnification.

An 8.0-cm MEN-1 pancreatic neuroendocrine tumor displaying biphasic morphology and heterogeneous immunolabeling for DAXX. The more typical pancreatic neuroendocrine morphology is seen on the left; a second morphologic pattern with prominent nucleoli and abundant cytoplasm is seen on the right. Both areas demonstrated the alternative lenghthening of the telomeres phenotype. H&E (a), DAXX (b). All images were taken at × 400 magnification.
Overall, 12 pancreatic neuroendocrine tumors measured ≥3 cm, and thus 25% (3 of 12) demonstrated loss of ATRX and/or DAXX and the presence of the alternative lengthening of telomeres phenotype (Table 2). Those with loss of ATRX and/or DAXX and the alternative lengthening of telomeres phenotype were more likely to be WHO grade 2 (as opposed to grade 1) based on KI67 immunolabeling than those without (3/3 vs 1/9, P=0.0182). The average size of the 12 pancreatic neuroendocrine tumors measuring ≥3 cm was 4.2 cm (range 3.0–8.0 cm).
Discussion
Understanding the timing of genetic alterations in tumor development can often help define their roles in progression of disease, and this has important implications for detection and treatment. Unlike the well-defined sequence from adenoma to invasive carcinoma in the colorectum, the histological progression of pancreatic neuroendocrine tumors is not well defined because no clear-cut basement membrane separates noninvasive from invasive lesions. Tumor size can serve as a surrogate for progression of pancreatic neuroendocrine tumors, as the vast majority of small lesions (ie, microadenomas) behave in a benign fashion, while larger lesions (ie, pancreatic neuroendocrine tumors, by definition measuring ≥0.5 cm) demonstrate malignant potential.16, 22, 23
In this study, we found that loss of nuclear expression of ATRX and/or DAXX occurred in 6% of MEN-1 well-differentiated pancreatic neuroendocrine tumors and that loss of nuclear expression of ATRX and/or DAXX perfectly correlated with the presence of the alternative lengthening of telomeres phenotype. These findings establish the existence of ATRX and DAXX defects and the alternative lengthening of telomeres telomere maintenance mechanism in the setting of MEN-1 syndrome pancreatic neuroendocrine tumors and confirm the correlation between loss of nuclear expression of ATRX or DAXX and the occurrence of the alternative lengthening of telomeres phenotype.
We also found that loss of nuclear expression of ATRX and/or DAXX and the presence of the alternative lengthening of telomeres phenotype occurred only in larger (≥3 cm) pancreatic neuroendocrine tumors in patients with MEN-1 syndrome. This finding suggests that these changes are ‘late’ events in the pancreatic neuroendocrine tumorigenesis, occurring only after the neoplasms have grown well beyond the size of microadenomas.
As compared with the prior study of sporadic pancreatic neuroendocrine tumors by Jiao et al,2 the prevalence of ATRX and/or DAXX defects and the alternative lengthening of telomeres phenotype in the current study of MEN-1 tumors was considerably less (43% vs 6%). While inherent differences between these two groups of neuroendocrine tumors may certainly be responsible for this discrepancy, it may also reflect differences in the size (ie, progression) of the pancreatic neuroendocrine tumors studied: the mean size of pancreatic neuroendocrine tumors in the prior study was 4.9 cm, whereas the mean size in the current study was 1.9 cm. Even within the subset of MEN-1 pancreatic neuroendocrine tumors measuring ≥3 cm in the current study (albeit a sample size too small for definitive conclusions), the mean size was just 4.2 cm, and notably, the proportion of these tumors showing ATRX and/or DAXX defects and the alternative lengthening of telomeres phenotype was 25% (a figure much closer to the 43% seen in the sporadic pancreatic neuroendocrine tumors). Additionally, it is worth noting that MEN1 mutation per se does not preclude an ATRX or DAXX defect, as nearly one-quarter of the sporadic pancreatic neuroendocrine tumors in the previous study harbored dual mutations in MEN1 and either ATRX or DAXX.2
Although the vast majority of neoplasms maintain their telomere lengths by increased activity of telomerase, ∼4% of neoplasms maintain their telomere lengths through a homologous recombination-based mechanism known as alternative lengthening of telomeres.17 One of the hallmarks of this telomere maintenance mechanism is accumulation of large amounts of telomeric DNA in discrete nuclear foci, a feature that is the basis for the telomere-specific FISH assay used to detect alternative lengthening of telomeres. In all tumors tested to date, including those of the current study, loss of nuclear ATRX and/or DAXX shows a near 100% correlation with the alternative lengthening of telomeres phenotype,3, 4 suggesting that inactivation of these proteins (and/or their associated genes) is a critical step in the development of this cancer-associated telomere maintenance mechanism. Although the clinical significance of these changes in pancreatic neuroendocrine tumors is still being unraveled, mutations in either ATRX or DAXX have been shown to be associated with improved survival (as compared with pancreatic neuroendocrine tumors without ATRX or DAXX mutations).2 Unfortunately, in the current study, the prevalence of ATRX and/or DAXX defects was too low to generate a statistically well-powered survival curve.
In summary, our findings establish the existence of ATRX and DAXX defects and the alternative lengthening of telomeres telomere maintenance mechanism in the setting of MEN-1 syndrome pancreatic neuroendocrine tumors, confirm the correlation between loss of nuclear ATRX or DAXX expression and the occurrence of the alternative lengthening of telomeres phenotype, and demonstrate that these changes occur as late events in pancreatic neuroendocrine tumor development in patients with MEN-1 syndrome. Furthermore, the presence of simultaneous defects in ATRX or DAXX and MEN1 in this study adds to a growing body of evidence that the signaling pathway of ATRX and DAXX is distinct from that of MEN1.2, 24, 25, 26, 27, 28, 29, 30
References
Hochwald SN, Zee S, Conlon KC, et al. Prognostic factors in pancreatic endocrine neoplasms: An analysis of 136 cases with a proposal for low-grade and intermediate-grade groups. J Clin Oncol 2002;20:2633–2642.
Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011;331:1199–1203.
Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011;333:425.
Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012;484:226–231.
Anlauf M, Schlenger R, Perren A, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol 2006;30:560–574.
Perren A, Anlauf M, Henopp T, et al. Multiple endocrine neoplasia type 1 (MEN1): Loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab 2007;92:1118–1128.
Eubanks PJ, Sawicki MP, Samara GJ, et al. Putative tumor-suppressor gene on chromosome 11 is important in sporadic endocrine tumor formation. Am J Surg 1994;167:180–185.
Debelenko LV, Zhuang Z, Emmert-Buck MR, et al. Allelic deletions on chromosome 11q13 in multiple endocrine neoplasia type 1-associated and sporadic gastrinomas and pancreatic endocrine tumors. Cancer Res 1997;57:2238–2243.
Zhuang Z, Vortmeyer AO, Pack S, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 1997;57:4682–4686.
Wang EH, Ebrahimi SA, Wu AY, et al. Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Res 1998;58:4417–4420.
Hessman O, Lindberg D, Skogseid B, et al. Mutation of the multiple endocrine neoplasia type 1 gene in nonfamilial, malignant tumors of the endocrine pancreas. Cancer Res 1998;58:377–379.
Hessman O, Lindberg D, Einarsson A, et al. Genetic alterations on 3p, 11q13, and 18q in nonfamilial and MEN 1-associated pancreatic endocrine tumors. Genes Chromosomes Cancer 1999;26:258–264.
Gortz B, Roth J, Krahenmann A, et al. Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol 1999;154:429–436.
Moore PS, Missiaglia E, Antonello D, et al. Role of disease-causing genes in sporadic pancreatic endocrine tumors: MEN1 and VHL. Genes Chromosomes Cancer 2001;32:177–181.
D'adda T, Pizzi S, Azzoni C, et al. Different patterns of 11q allelic losses in digestive endocrine tumors. Hum Pathol 2002;33:322–329.
Klimstra DS, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the pancreas. In: Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 4th edn. WHO Press: Geneva, Switzerland, 2010, pp 322–326.
Heaphy CM, Subhawong AP, Hong SM, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 2011;179:1608–1615.
Marciniak RA, Cavazos D, Montellano R, et al. A novel telomere structure in a human alternative lengthening of telomeres cell line. Cancer Res 2005;65:2730–2737.
Fasching CL, Bower K, Reddel RR . Telomerase-independent telomere length maintenance in the absence of alternative lengthening of telomeres-associated promyelocytic leukemia bodies. Cancer Res 2005;65:2722–2729.
Cerone MA, Autexier C, Londono-Vallejo JA, et al. A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene 2005;24:7893–7901.
Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Thiese ND (eds). WHO classification of tumours of the digestive system. 4th edn. WHO Press: Geneva, Switzerland, 2010, pp 13–14.
Modlin IM, Sandor A . An analysis of 8305 cases of carcinoid tumors. Cancer 1997;79:813–829.
Yao JC, Hassan M, Phan A, et al. One hundred years after "carcinoid": Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the united states. J Clin Oncol 2008;26:3063–3072.
Xue Y, Gibbons R, Yan Z, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA 2003;100:10635–10640.
Yokoyama A, Wang Z, Wysocka J, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 2004;24:5639–5649.
Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol Cell 2004;13:587–597.
Wong LH, Ren H, Williams E, et al. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res 2009;19:404–414.
Wong LH, McGhie JD, Sim M, et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 2010;20:351–360.
Lewis PW, Elsaesser SJ, Noh KM, et al. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA 2010;107:14075–14080.
Drane P, Ouararhni K, Depaux A, et al. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 2010;24:1253–1265.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
de Wilde, R., Heaphy, C., Maitra, A. et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol 25, 1033–1039 (2012). https://doi.org/10.1038/modpathol.2012.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.53
Keywords
This article is cited by
-
Molecular Pathology of Well-Differentiated Gastro-entero-pancreatic Neuroendocrine Tumors
Endocrine Pathology (2021)
-
Centromeric cohesion failure invokes a conserved choreography of chromosomal mis-segregations in pancreatic neuroendocrine tumor
Genome Medicine (2020)
-
Liver metastases from pituitary carcinomas mimicking visceral well-differentiated neuroendocrine tumors: a series of four cases
Diagnostic Pathology (2020)
-
Morphologic Variants of Pancreatic Neuroendocrine Tumors: Clinicopathologic Analysis and Prognostic Stratification
Endocrine Pathology (2020)
-
Alternative Lengthening of Telomeres and Differential Expression of Endocrine Transcription Factors Distinguish Metastatic and Non-metastatic Insulinomas
Endocrine Pathology (2020)
