
Abstract
Benign changes ranging from atrophy and inflammation to high-grade prostatic intraepithelial neoplasia (HGPIN) are common findings on prostate core needle biopsies. Although atrophy and inflammation may be precursors of prostate cancer, only HGPIN is currently recommended to be included in surgical pathology reports. To determine whether these benign findings increase prostate cancer risk, we conducted a case–control study nested within a historical cohort of 6692 men with a benign prostate specimen collected between 1990 and 2002. The analytic sample included 574 case–control pairs comprised of cases diagnosed with prostate cancer a minimum of 1 year after cohort entry and controls matched to cases on date and age at cohort entry, race, and type of specimen. The initial benign specimen was reviewed for presence of HGPIN, atrophy (simple, lobular, and partial) and inflammation (glandular and/or stromal). HGPIN significantly increased risk for prostate cancer (odds ratio (OR)=2.00; 95% confidence interval (CI)=1.25–3.20). Inflammation within the stromal compartment was associated with decreased risk (OR=0.66; CI=0.52–0.84), and diffuse stromal inflammation of severe grade had the strongest inverse association with risk (OR=0.21; CI=0.07–0.62). In a model adjusted for prostate-specific antigen (PSA) level at cohort entry and inflammation, simple atrophy was associated with a 33% increased prostate cancer risk that was marginally significant (P=0.03). Clinicians should consider patterns and extent of inflammation when managing high-risk patients with negative biopsy results. Identifying benign inflammatory processes that underlie high PSA levels would help to reduce the number of unnecessary repeated prostate biopsies.
Similar content being viewed by others
Main
Benign changes ranging from atrophy and inflammation to prostatic intraepithelial neoplasia are common findings on prostate needle core biopsies.1, 2, 3, 4 Of these, only high-grade prostatic intraepithelial neoplasia (HGPIN) is routinely recorded on pathologic reports because of its well-known association with prostate cancer.5, 6, 7 One study reports that 77% of urologists consider the presence of HGPIN in the absence of cancer to be an indication for subsequent rebiopsy,8 and the need for rebiopsy after benign biopsy with evidence of HGPIN has been suggested by several pathology-based studies.9, 10, 11
The observation that foci of prostatic carcinoma and HGPIN are often associated with foci of simple atrophy has led to the suggestion that atrophy may be involved in the initiation of prostate cancer.12, 13 McNeal14 used the term ‘post-inflammatory atrophy’ to characterize the association of simple atrophy with chronic inflammation. De Marzo et al15, 16 introduced the term ‘proliferative inflammatory atrophy’ for such lesions and suggested that they may be precursors to HGPIN and cancer. This nomenclature implies that inflammation is the cause of atrophy; however, foci of simple atrophy demonstrate a high proliferation index when compared with normal prostatic epithelium even in the absence of inflammation.17, 18, 19
Despite the biologic plausibility of proliferative inflammatory atrophy being a precursor of carcinoma, the majority of evidence for this theory comes from the proximity of such lesions to cancer in prostatectomy specimens.12, 13 While focal atrophy has been considered a feature of the aging prostate gland,1 such changes were found in as many as 70% of young individuals,4, 20 with incidence ranging from 88 to 100% in older individuals.1, 2, 4, 21 Given the high frequency of atrophy, and the tendency of both HGPIN and atrophy to locate in the peripheral zone of the prostate, the relationship between proliferative inflammatory atrophy and cancer may be circumstantial.
Likewise, inflammation is often histologically apparent in the examination of prostate specimens from older men. The cause of chronic prostatic inflammation as well as its putative role in carcinogenesis remain unclear.22 Previous studies have found both positive23 and negative24 associations between inflammation and incidence of prostate cancer. The positive correlation between inflammation and circulating prostate-specific antigen (PSA) levels25, 26, 27 strengthens the biologic plausibility of an inflammation–cancer link, but also confounds studies aimed at determining whether an association exists between the two. In patients with a negative biopsy performed due to elevated PSA, either undetected cancer or subclinical prostatitis (if histologic evidence of inflammation is present) may serve as plausible explanations for the high PSA level. Improving the characterization of the types of inflammation that indicate a benign process vs an early stage cancer remains a challenge.
Atrophy and inflammation are the most frequent benign prostate biopsy findings, and may be precursors of prostate cancer. But what—if any—risk is associated with these benign lesions has not been well established. To date, our work represents the largest case–control study in an ethnically diverse population to examine whether benign prostate lesions are risk factors for prostate cancer.
Materials and methods
Study Sample
After obtaining appropriate approval from the Henry Ford Health System Institutional Review Board, we identified a historical cohort of 6692 men with a benign prostate specimen collected by needle core biopsy or transurethral resection of the prostate between January 1990 and December 2002. Within this cohort, a nested case–control sample was assembled. Eligibility criteria included a recorded PSA level within a year of cohort entry and no history of a previous prostate cancer diagnosis. ‘Date of cohort entry’ was defined as the date of initial benign prostate biopsy; ‘date of case diagnosis’ was the date of first cancer-positive tissue specimen or the date a clinician first reported a clinical diagnosis of prostate cancer. Patients diagnosed with prostate cancer <1 year from date of initial biopsy were ineligible for the study. We identified 808 potentially eligible cases diagnosed with prostate cancer before July 2007.
Incidence density sampling was used to select controls with replacement from all cohort members at risk at the time of case occurrence. Controls were randomly selected from among those cohort members who were free of prostate cancer at a follow-up duration greater than or equal to the time between cohort entry and diagnosis of the matched case. Matching criteria included age at entry into cohort (±2 years), date of entry into cohort (±2 years), race (African American or White), and type of specimen (biopsy or transurethral resection). We were able to match 802 of 808 potentially eligible cases. Further review reduced the final analytic sample to 574 case–control pairs. Exclusions were primarily due to problems with tissue blocks (n=126; 55%), including lack of analyzable prostate tissue, wrong specimen type, or missing specimens. Other exclusions included absence of a PSA test within 1 year of cohort entry (n=37; 16%) and evidence of malignancy (n=29; 13%) after a second pathologic review of specimens initially characterized as benign. Further medical record review found earlier benign specimens (outside the cohort window) that made 12 (5%) pairs ineligible. The remaining pairs (n=24; 10%) were excluded for reasons related to incomplete records at the time of cohort entry or diagnosis.
Pathologic Review
All benign tissue specimens were evaluated for the presence of cancer, HGPIN, atrophy, and inflammation by a single genitourinary pathologist (ONK) blinded to disease progression. HGPIN was defined using criteria defined by McNeal and Bostwick; all HGPIN lesions shared similar characteristic cytological features regardless of the pattern of presentation.5, 6, 28 Differentiation between ambiguous high- or low-grade PIN was made on the basis of mitotic activity as proposed by Epstein and Netto.29 Atrophy was categorized following the system developed by De Marzo et al1 (simple atrophy; post-atrophic hyperplasia; simple atrophy—cyst formation; partial atrophy; Figure 1). The location (acinar, periacinar, stromal) and grade (mild, moderate, severe) of inflammation was assessed according to the criteria proposed by Nickel et al3 (Figure 2). For cases with varying intensity of inflammatory infiltrate, the highest grade was recorded. The extent of HGPIN, atrophy, and inflammation was scored as focal (<10% of specimen), multifocal (10–50% of specimen), or diffuse (>50% of specimen). Specimens with evidence of non-reported malignancy were reviewed by a second pathologist (DAC) before exclusion from the sample.

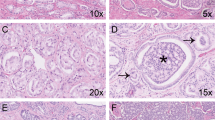
(a) Simple atrophy in needle biopsy core. Note presence of diffuse stromal, periglandular, and glandular inflammation. Glandular inflammation was consistently associated with atrophic acini and ducts. (b) Simple atrophy continuous with partial atrophy. (c) Simple atrophy partially involving the prostatic acinus. Inflammation was not present in foci where simple atrophy involved only part of the acinus or duct.

(a) Diffuse severe stromal inflammation. Periglandular and stromal inflammation was associated with foci of simple atrophy, particularly when consecutive acini and ducts were involved. (b) Focus of severe stromal inflammation (higher magnification). Note the distance between the inflammatory aggregate and surrounding atrophic acini. (c) Severe periglandular inflammation. (d) Glandular inflammation in the atrophic gland. Note the presence of intraluminal and intraepithelial inflammatory cells.
Statistical Analysis
Conditional logistic regression analyses were used to estimate odds ratios (ORs) for prostate cancer incidence during follow-up. Individual matching controlled for age, race, and specimen type. Initial univariate models assessed associations between prostate cancer incidence and the following: presence of HGPIN; presence of any atrophy and subtypes of atrophy; and presence of any inflammation and inflammation by location, by grade, and by extent. Next, multivariable models were fitted to include variables for PSA level at benign biopsy, presence of HGPIN, presence of any atrophy, and presence of any inflammation. Subsequent multivariable models considered the presence of subtypes of atrophy and inflammation by location, by grade, and by extent. Lastly, a series of stratified multivariable models were fit to include variables for PSA level at benign biopsy, presence of HGPIN, and presence of stromal atrophy, with models stratified on age (based on median age at cohort entry), race, median time to diagnosis, and median date of cohort entry. Comparisons between the stratified models were assessed using a conditional logistic regression model that included interaction terms with the stratified variable.
Results
Table 1 shows the demographic and clinical characteristics of the study sample that comprised 574 matched case–control pairs. Most benign specimens analyzed were needle core biopsies (94%). Cases were 40% African American with an average age of 66 years at cohort entry. Date of cohort entry ranged from 1990 to 2002; the median date of cohort entry was February 1995 for cases and March 1995 for controls. By design, cases were diagnosed with cancer at least 1 year post-cohort entry; the median time to cancer diagnosis was 4 years, with some cases diagnosed up to 15 years after cohort entry. Cases had a significantly higher PSA level at the time of cohort entry, higher PSA velocity from the time of cohort entry to diagnosis or to control matching, and averaged two more PSA tests within this time period. The majority of cases were diagnosed with stage 1 (39%) and 2 (52%) tumors. Approximately 30% of cases had advanced tumor grade, defined as either Gleason score 7 with a primary grade 4, or 8 and above.
The prevalence of HGPIN, focal atrophy, and inflammation in the initial benign prostate samples of both cases and controls was examined first (Table 2). Presence of HGPIN was associated with a twofold increased risk for prostate cancer (OR=2.00; 95% confidence interval (CI)=1.25–3.20). Simple atrophy and post-atrophic hyperplasia were both found more frequently in case samples, but neither was significantly associated with case status. Likewise, the presence of any atrophy was associated with a 24% increased risk for prostate cancer, but the results were not statistically significant (P=0.07).
The association between presence, location, grade, and extent of inflammation with risk of prostate cancer is shown in Table 3. The presence of any inflammation was associated with a statistically significant decreased risk for prostate cancer (OR=0.65; CI=0.51–0.84). This association was confined to inflammation within the stromal compartment (OR=0.66; CI=0.52–0.84); inflammation in glandular or periglandular regions was not associated with cancer risk. Grade and extent of inflammation was inversely associated with risk. For example, moderate (OR=0.55) and severe inflammation (OR=0.63) were more strongly inversely associated with prostate cancer than mild inflammation (OR=0.70). Likewise, diffuse inflammation (OR=0.40) had a stronger inverse association with prostate cancer than either focal (OR=0.68) or multifocal inflammation (OR=0.65).
Conversely, a positive correlation was observed between PSA level and presence of inflammation (Figures 3a–c). The presence of any inflammation was strongly associated with higher PSA in both cases and controls (Figure 3a). With the exception of glandular inflammation in controls, PSA levels were higher in the presence of inflammation irrespective of location. While the grade of inflammation did not correlate with PSA levels (Figure 3b), extent of inflammation—particularly in cases—was associated with increasing PSA levels (Figure 3c).

(a) Estimated mean PSA levels (ng/ml) and 95% confidence intervals by presence of inflammation and case/control status. (b) Estimated mean PSA levels (ng/ml) and 95% confidence intervals by inflammation grade and case/control status. (c) Estimated mean PSA levels (ng/ml) and 95% confidence intervals by inflammation extent and case/control status.
Next, a series of multivariable models were fit to determine the independent effects of atrophy and inflammation on prostate cancer risk (Table 4). These models included PSA levels and presence of HGPIN at cohort entry, both strongly associated with cancer risk. In the initial model, subtypes of atrophy and inflammation were compressed into two variables: presence of atrophy and presence of inflammation. As previously observed in the univariate analyses, presence of any inflammation had a strong inverse association with prostate cancer risk (P<0.001), whereas atrophy was associated with an ∼30% increased risk (P=0.04). When subtypes of atrophy were modeled; only simple atrophy was significantly associated with risk (OR=1.32; CI=1.02–1.67). Modeling the loci of inflammation (stromal, glandular, or periglandular) as separate variables and adjusting for PSA, HGPIN, and atrophy, the results were comparable to the univariate analyses, with stromal inflammation showing a strong inverse association with prostate cancer risk (P<0.001).
Finally, we also examined the joint effects of PSA and inflammation on prostate cancer risk. The interaction OR of PSA and presence of any inflammation was <1 (OR=0.86; P=0.0001) and highly statistically significant, indicative of an antagonistic effect between the two variables with regard to prostate cancer risk. For example, at a PSA level of 10 ng/ml, the OR for inflammation was 0.33 (CI=0.22–0.50), whereas at a PSA level of 4 ng/ml, the OR associated with inflammation was 0.77 (CI=0.58–1.03). The interaction OR for PSA and stromal inflammation was also <1 (OR=0.95; P=0.051), but only marginally statistically significant, suggesting that PSA levels were less of an effect modifier for this risk variable.
To further investigate this relationship between stromal inflammation and decreased prostate cancer risk, a model was created to include additional variables for combined extent and grade of stromal inflammation (Table 5). There was a general downward trend of ORs with combinations of increased extent and grade of inflammation. Men with diffuse and severe stromal inflammation had the lowest risk of prostate cancer compared with men without stromal inflammation (OR=0.20; CI=0.07–0.60).
Table 6 shows results of analyses for the effects of HGPIN, stromal inflammation, and simple atrophy across strata of age, race, time, and tumor grade. There was a suggestion of HGPIN being more strongly associated with prostate cancer risk in younger men (OR=2.89 vs 1.89), but differences between age strata were not statistically significant (P=0.5). The risk associated with HGPIN was stronger for cancer diagnosed 1–4 years after cohort entry than for cancer diagnosed >4 years after cohort entry. Risk of prostate cancer was also significantly greater (P=0.03) for men with HGPIN who entered the cohort later (after March 1995) vs earlier. Further investigation of the relationship between time of cohort entry, time to diagnosis, and HGPIN revealed a potential synergistic effect. HGPIN was associated with a sevenfold increase in risk (OR=7.33; CI=1.98–27.15) in men who entered the cohort late and who had cancer diagnosed 1–4 years after cohort entry, whereas for men who entered the cohort early and who had cancer diagnosed ≥4 years after entry, HGPIN had no effect on prostate cancer risk (OR=0.63; CI=0.24–1.64). No notable differences in effect estimates for either HGPIN or stromal inflammation were observed between the race and tumor grade strata.
Discussion
This study confirms previously reported associations of HGPIN with increased risk of subsequent prostate cancer diagnosis.5, 6, 7 In this nested cohort, the presence of HGPIN was associated with a twofold increased risk for prostate cancer. Overall, prostate cancer was diagnosed in 65% (56/86) of patients with HGPIN present in their initial benign prostate specimen, the majority being diagnosed within 4 years of follow-up. While Davidson et al30 reported a relative risk of 14.93 for prostate cancer associated with HGPIN, that study included only 3 years of follow-up, with most cases diagnosed in the first year. Subsequent studies have estimated a twofold to fourfold increase in risk for prostate cancer following a diagnosis of HGPIN.31, 32, 33 In the present study, the prostate cancer risk associated with the presence of HGPIN was strongest for cancer diagnosed 1–4 years after cohort entry (OR=5.68; CI=2.18–13.71), with the risk for HGPIN also greater in the subset who entered the cohort after March 1995 (OR=3.62; CI=1.73–7.56). Interestingly, analyzing case–control pairs that fell into both of these two subsets (those diagnosed 1–4 years after cohort entry and entering the cohort after March 1995) resulted in an even greater cancer risk associated with HGPIN (OR=7.33; CI=1.98–27.15, not shown in Table). These results suggest that cancer arising from HGPIN does so in a short time period and that current intensive screening practices (reflected best in the later half of our cohort) may further accentuate this process. Evidence from a large cohort study exists that extent of HGPIN on biopsy is a strong risk factor for subsequent prostate cancer diagnosis in men with an initial benign biopsy.32, 33 In our study, we did not extensively examine all benign samples of each study participant, but rather representative tissue samples. In these samples, over 90% of the HGPIN-positive samples had focal HGPIN, which precluded any meaningful analysis of extent of HGPIN as a prostate cancer risk factor.
The relationship between atrophy and prostate cancer is less straightforward. Over the last two decades, speculation has arisen that atrophy and inflammation may be harbingers of prostate carcinogenesis.12, 13, 16, 22, 34 Foci of simple atrophy can be adjacent to or intermixed with cancer (Figure 4), and while proliferative inflammatory atrophy may have a role in prostate carcinogenesis, neither atrophy in areas of cancer nor cancer itself are associated with any type of inflammation. In this study, simple atrophy was associated with 20–30% increased risk for prostate cancer that attained statistical significance after adjusting for presence of HGPIN and PSA at baseline. Only two studies have prospectively tested the association between atrophic lesions on biopsy and a subsequent prostate cancer diagnosis. The prevalence of simple atrophy (43%) in the present study was similar to that reported by Asimakopoulos et al35 in 351 benign biopsy specimens. In 202 randomly selected cases from the European Randomized Study of Screening for Prostate Cancer, Postma et al4 found that neither the subtype nor extent of atrophy was significantly associated with a subsequent diagnosis of prostate cancer. Other cross-sectional reports generally fail to find a definitive association between atrophy and prostate cancer.2, 21, 36 Our results suggest that patients with atrophy observed in a benign prostate biopsy are at a modestly increased risk for prostate cancer and therefore may warrant more vigilant follow-up.

(a) Gland with simple atrophy surrounded by prostate cancer. (b) Intraductal spread of prostate cancer. Note atrophic native epithelium at the periphery of the duct and markedly enlarged and pleomorphic nuclei of intraductal carcinoma.
Like atrophy, the relationship between inflammation and prostate cancer remains unclear.15 Previous studies have generally investigated the relationship of inflammation with atrophy or with PSA. For example, MacLennan et al23 used a pathology database to investigate the role of inflammation; while they showed a much higher incidence of prostate cancer at 5 years of follow-up in cases with chronic inflammation at initial biopsy, case ascertainment issues and modest sample size limit inference from their findings. Wolters et al37 analyzed HGPIN and inflammation in 121 patients who had benign prostate biopsies followed by rebiopsy 4 years later; neither was predictive of cancer risk. Terakawa et al24 reported that inflammation is more often observed in benign prostatic disease; in a cross-sectional study of 143 consecutive patients, absence of chronic inflammation in the prostate was associated with a threefold increased risk for prostate cancer.
In our study, inflammation—specifically stromal inflammation—was associated with a decreased risk of prostate cancer. This finding remained after adjustment for PSA levels at cohort entry and presence of HGPIN, and became stronger with increasing extent of inflammation. It is unclear whether this apparent protective effect reflects a biological process or a selection bias due to the association between inflammation and PSA. Inflammation is associated with elevated serum PSA levels, which brings patients to urologists' attention and increases the likelihood of biopsy and thus, entrance into a screened cohort.26, 27 If inflammation-associated increase in PSA does not reflect a carcinogenic process, the presence of inflammation will select men into the cohort who are at average or perhaps slightly elevated risk of prostate cancer. Thus, when these men with inflammation are compared with the other high risk men in the cohort, they will appear to have a lower incidence of prostate cancer during follow-up. This is probably best illustrated in the negative interaction OR we found between inflammation and PSA, which suggests that inflammation is only a protective factor for prostate cancer at high PSA levels. That is, men with high PSA and inflammation are at lower risk for prostate cancer than men with high PSA and no inflammation.
The findings of our study may not be generalizable to all men. By definition, cohort members were candidates for surgical prostate interventions, and therefore cannot be considered representative of all men in the same age and race demographic. We excluded patients diagnosed with cancer within a year after cohort entry to minimize the chance of undetected prostate cancer. Nonetheless, based on the age range of our cohort and the high prevalence of undiagnosed prostate cancer in older men,38 some men in our cohort likely had synchronous prostate cancer that was missed on initial biopsy. Since cohort members were followed for up to 15 years, we were able to make long-term estimates of risk that are less likely to be biased by contamination of the cohort with undiagnosed prostate cancer.39 Our study matched on several factors that allowed us to control for temporal changes in prostate screening and detection that occurred between 1990 and 2007. The age distribution of our study population was comparable with typical age of prostate cancer diagnosed in the United States during this time.40 Cases tended to have tumors of more advanced stage and grade than might be observed in a random series, again reflecting how the initial cohort was ascertained. Despite the inherent shortcomings of a retrospective cohort design, embedding the cohort within a single health system permitted efficient sampling and complete incident case detection. This allowed accurate estimation of temporal associations between benign lesions of the prostate and subsequent risk of cancer. In summary, our study cohort is likely to reflect the risk profiles of men who have been biopsied and found to have benign conditions but remain under medical surveillance.
In summary, our results suggest that simple atrophy may modestly increase risk of prostate cancer whereas presence of stromal inflammation significantly decreases risk. Moreover, characterizing the type and extent of inflammation may explain the main cause of elevated PSA in patients with negative prostate biopsy. Such characterization would aid in the clinical management of these patients, by reducing the number of unnecessary repeated prostate biopsies.
References
De Marzo AM, Platz EA, Epstein JI, et al. A working group classification of focal prostate atrophy lesions. Am J Surg Pathol 2006;30:1281–1291.
Billis A, Magna LA . Inflammatory atrophy of the prostate. Prevalence and significance. Arch Pathol Lab Med 2003;127:840–844.
Nickel JC, True LD, Krieger JN, et al. Consensus development of a histopathological classification system for chronic prostatic inflammation. BJU Int 2001;87:797–805.
Postma R, Schroder FH, van der Kwast TH . Atrophy in prostate needle biopsy cores and its relationship to prostate cancer incidence in screened men. Urology 2005;65:745–749.
McNeal JE, Bostwick DG . Intraductal dysplasia: a premalignant lesion of the prostate. Hum Pathol 1986;17:64–71.
Bostwick DG, Qian J . High-grade prostatic intraepithelial neoplasia. Mod Pathol 2004;17:360–379.
Epstein JI, Herawi M . Prostate needle biopsies containing prostatic intraepithelial neoplasia or atypical foci suspicious for carcinoma: implications for patient care. J Urol 2006;175:820–834.
Descazeaud A, Rubin MA, Allory Y, et al. What information are urologists extracting from prostate needle biopsy reports and what do they need for clinical management of prostate cancer? Eur Urol 2005;48:911–915.
De Matteis M, Poggi C, De Martino A, et al. Repeat biopsy in patients with initial diagnosis of PIN. Radiol Med 2005;110:190–198.
Borboroglu PG, Sur RL, Roberts JL, et al. Repeat biopsy strategy in patients with atypical small acinar proliferation or high grade prostatic intraepithelial neoplasia on initial prostate needle biopsy. J Urol 2001;166:866–870.
Netto GJ, Epstein JI . Widespread high-grade prostatic intraepithelial neoplasia on prostatic needle biopsy: a significant likelihood of subsequently diagnosed adenocarcinoma. Am J Surg Pathol 2006;30:1184–1188.
Putzi MJ, De Marzo AM . Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia. Urology 2000;56:828–832.
Wang W, Bergh A, Damber JE . Morphological transition of proliferative inflammatory atrophy to high-grade intraepithelial neoplasia and cancer in human prostate. Prostate 2009;69:1378–1386.
McNeal JE . Normal histology of the prostate. Am J Surg Pathol 1988;12:619–633.
De Marzo AM, Marchi VL, Epstein JI, et al. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol 1999;155:1985–1992.
De Marzo AM, Nakai Y, Nelson WG . Inflammation, atrophy, and prostate carcinogenesis. Urol Oncol 2007;25:398–400.
Liavag I . Mitotic activity of prostatic epithelium. A study by means of Colcemid. Acta Pathol Microbiol Scand 1968;73:19–28.
Ruska KM, Sauvageot J, Epstein JI . Histology and cellular kinetics of prostatic atrophy. Am J Surg Pathol 1998;22:1073–1077.
Feneley MR, Young MP, Chinyama C, et al. Ki-67 expression in early prostate cancer and associated pathological lesions. J Clin Pathol 1996;49:741–748.
Gardner Jr WA, Culberson DE . Atrophy and proliferation in the young adult prostate. J Urol 1987;137:53–56.
Tomas D, Kruslin B, Rogatsch H, et al. Different types of atrophy in the prostate with and without adenocarcinoma. Eur Urol 2007;51:98–103; discussion 103–104.
De Marzo AM, Platz EA, Sutcliffe S, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer 2007;7:256–269.
MacLennan GT, Eisenberg R, Fleshman RL, et al. The influence of chronic inflammation in prostatic carcinogenesis: a 5-year followup study. J Urol 2006;176:1012–1016.
Terakawa T, Miyake H, Kanomata N, et al. Inverse association between histologic inflammation in needle biopsy specimens and prostate cancer in men with serum PSA of 10-50 ng/mL. Urology 2008;72:1194–1197.
International consultation on prostatic intraepithelial neoplasia and pathologic staging of prostatic carcinoma. Rochester, Minnesota, November 3-4, 1995. Cancer 1996;78:320–381.
Schatteman PH, Hoekx L, Wyndaele JJ, et al. Inflammation in prostate biopsies of men without prostatic malignancy or clinical prostatitis: correlation with total serum PSA and PSA density. Eur Urol 2000;37:404–412.
Stimac G, Reljic A, Spajic B, et al. Aggressiveness of inflammation in histological prostatitis--correlation with total and free prostate specific antigen levels in men with biochemical criteria for prostate biopsy. Scott Med J 2009;54:8–12.
Bostwick DG, Amin MB, Dundore P, et al. Architectural patterns of high-grade prostatic intraepithelial neoplasia. Hum Pathol 1993;24:298–310.
Epstein JI, Netto GJ . Prostatic and intraepithelial neoplasia and its mimickers. In: Epstein JI (ed). Biopsy Interpretation of the Prostate 4th edn. LWW: Philadelphia, 2008, pp 35–67.
Davidson D, Bostwick DG, Qian J, et al. Prostatic intraepithelial neoplasia is a risk factor for adenocarcinoma: predictive accuracy in needle biopsies. J Urol 1995;154:1295–1299.
Lee MC, Moussa AS, Yu C, et al. Multifocal high grade prostatic intraepithelial neoplasia is a risk factor for subsequent prostate cancer. J Urol 2010;184:1958–1962.
Merrimen JL, Jones G, Walker D, et al. Multifocal high grade prostatic intraepithelial neoplasia is a significant risk factor for prostatic adenocarcinoma. J Urol 2009;182:485–490; discussion 490.
Merrimen JL, Jones G, Srigley JR . Is high grade prostatic intraepithelial neoplasia still a risk factor for adenocarcinoma in the era of extended biopsy sampling? Pathology 2010;42:325–329.
Palapattu GS, Sutcliffe S, Bastian PJ, et al. Prostate carcinogenesis and inflammation: emerging insights. Carcinogenesis 2005;26:1170–1181.
Asimakopoulos AD, Miano R, Mauriello A, et al. Significance of focal proliferative atrophy lesions in prostate biopsy cores that test negative for prostate carcinoma. Urol Oncol 2011;29:690–697.
Stamatiou K, Alevizos A, Agapitos E, et al. Incidence of impalpable carcinoma of the prostate and of non-malignant and precarcinomatous lesions in Greek male population: an autopsy study. Prostate 2006;66:1319–1328.
Wolters T, Roobol MJ, Schroder FH, et al. Can non-malignant biopsy features identify men at increased risk of biopsy-detectable prostate cancer at re-screening after 4 years? BJU Int 2008;101:283–288.
Sakr WA, Grignon DJ, Crissman JD, et al. High grade prostatic intraepithelial neoplasia (HGPIN) and prostatic adenocarcinoma between the ages of 20--69: an autopsy study of 249 cases. In Vivo 1994;8:439–443.
Nonn L, Ananthanarayanan V, Gann PH . Evidence for field cancerization of the prostate. Prostate 2009;69:1470–1479.
NIH. Surveillance Epidemiology and End Results. Stat Fact Sheets: Prostate.http://www.seer.cancer.gov/statfacts/html/prost.html 2011.
Acknowledgements
We thank the medical record abstractors and other study personnel that helped with data collection for this study. This work was supported by NIH Grant # 5R01-ES011126.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This project was presented in part at Unites States and Canadian Academy of Pathology annual meeting in 2011.
Rights and permissions
About this article
Cite this article
Kryvenko, O., Jankowski, M., Chitale, D. et al. Inflammation and preneoplastic lesions in benign prostate as risk factors for prostate cancer. Mod Pathol 25, 1023–1032 (2012). https://doi.org/10.1038/modpathol.2012.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.51
Keywords
This article is cited by
-
The prevention of 2,4-dinitrochlorobenzene-induced inflammation in atopic dermatitis-like skin lesions in BALB/c mice by Jawoongo
BMC Complementary and Alternative Medicine (2018)
-
Macrophage Cytokines Enhance Cell Proliferation of Normal Prostate Epithelial Cells through Activation of ERK and Akt
Scientific Reports (2018)
-
Racial differences in the relationship between clinical prostatitis, presence of inflammation in benign prostate and subsequent risk of prostate cancer
Prostate Cancer and Prostatic Diseases (2016)
-
Greater extent of prostate inflammation in negative biopsies is associated with lower risk of prostate cancer on repeat biopsy: results from the REDUCE study
Prostate Cancer and Prostatic Diseases (2016)
-
Inflammation and prostate cancer: friends or foe?
Inflammation Research (2015)
