
Abstract
Rhabdomyosarcoma is a rare soft tissue sarcoma that typically affects children, adolescents, and young adults. Despite treatment via a multidisciplinary approach, the prognosis of advance-stage rhabdomyosarcomas remains poor, and a new treatment strategy is needed. Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase that is a potential target for specific inhibitors. In this study, we investigated 116 rhabdomyosarcomas using a polymer-based ALK immunostaining method and correlated the results with clinicopathological parameters. In addition, we examined ALK status using dual-color fluorescence in situ hybridization, PCR, and sequencing. In immunohistochemical analysis, ALK was detected in 2 (6%) of 33 embryonal rhabdomyosarcomas, 42 (69%) of 61 alveolar rhabdomyosarcomas, and 0 (0%) of 22 other subtypes, including pleomorphic, adult-spindle-cell/sclerosing, and epithelioid variants. Compared with ALK-negative alveolar rhabdomyosarcomas, ALK-positive ones are presented with metastatic spread more frequently and showed a greater extent of myogenin reactivity. Overall survival was not associated with ALK expression. FOXO1 rearrangement was significantly associated with ALK immunoreactivity. The median ALK copy number was greater in ALK-positive tumors than in ALK-negative tumors. Most (93%) cases tested showed no selective increase in the ALK gene dosage. ALK selective amplification and low-level selective gain were noted in one and three cases, respectively. Further, a high-polysomy pattern (≥4 ALK copies in ≥40% of cells) was observed in seven cases. A significant increase in the ALK copy number was exclusive to the ALK-immunopositive cohort, but it was uncommon, accounting for only 30% of the 37 ALK-positive rhabdomyosarcomas. ALK gene rearrangement was not observed in either cohort, while an ALK somatic mutation (I1277T) was found in one ALK-negative embryonal case. Although it remains controversial whether ALK expression without gene rearrangement is therapeutically relevant, this comprehensive analysis may help future studies on the utility of ALK-targeted therapy for patients with rhabdomyosarcoma.
Similar content being viewed by others

Main
Rhabdomyosarcoma is a rare sarcoma with skeletal muscle differentiation that typically affects children, adolescents, and young adults. It is histologically classified into the embryonal, alveolar, and pleomorphic subtypes,1, 2, 3 although other rare variants like sclerosing, adult-spindle-cell, and epithelioid rhabdomyosarcomas have also been proposed.4, 5, 6, 7 Each subtype is characterized by a distinct epidemiology, clinical behavior, and genetic background.1, 2, 3 For example, alveolar rhabdomyosarcoma affects older patients compared with the embryonal subtype, is associated with poor survival, and in most cases, is characterized by a specific t(2;13) or t(1;13) translocation, which results in the PAX3-FOXO1 or PAX7-FOXO1 fusion gene, respectively.3 Although multidisciplinary treatment has dramatically improved prognosis, the survival rate of those affected by metastatic rhabdomyosarcoma remains low. Therefore, a new treatment strategy is needed.
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase whose gene ALK is located on chromosome 2. The ALK gene is rearranged in anaplastic large cell lymphoma,8 ALK-positive B-cell lymphoma,9 inflammatory myofibroblastic tumor,10 and a small subset of lung carcinoma11 and renal cell carcinoma,12, 13 while it is mutated in neuroblastoma14 and in rare thyroid cancers.15 The ALK protein is constitutively expressed in these ALK-associated cancers, whereby it has a critical oncogenic role. ALK has recently received significant clinical attention because it is targetable by small molecule kinase inhibitors. The ALK inhibitor crizotinib showed a marked overall response and good disease control rates in patients with ALK-rearranged lung cancers,16 and similar encouraging results were reported for this drug in the case of an inflammatory myofibroblastic tumor17 and anaplastic large cell lymphomas.18
Considering several reports that have documented ALK expression in rhabdomyosarcomas,19, 20, 21, 22, 23 we speculated that ALK may also have an oncogenic role in these sarcomas, and that they may be treatable with ALK inhibitors. We first decided to clarify the incidence of ALK expression and genomic changes in rhabdomyosarcomas in relation to clinicopathological parameters. We investigated ALK expression in a large number of cases using a previously validated ALK immunostaining method24 and correlated the results with the tumor subtype, FOXO1 rearrangement, and clinicopathological findings. In addition, we determined ALK gene status (rearrangement, changes in copy number, and somatic mutations) in these sarcomas.
Materials and methods
Case Selection
This study was approved by the institutional review board. We retrieved 116 rhabdomyosarcoma specimens, each from a unique patient, from the pathology files of the National Cancer Center Hospital and the University of Tokyo Hospital (both in Tokyo, Japan). All hematoxylin and eosin-stained sections were reviewed along with the relevant immunostaining data, and the diagnoses were confirmed. All the tumors were positive for desmin and one or both of the myoregulatory proteins (myogenin and myoD1).25 Of note, our study population predominantly included adults: 60% of the patients were ≥18 years of age (median age, 20 years). All the tumors were reclassified on the basis of the latest World Health Organization scheme1, 2, 3 and the recent literature,4, 5, 6, 7 into embryonal (n=33), alveolar (n=61), pleomorphic (n=8), adult-spindle-cell/sclerosing (n=12), and epithelioid rhabdomyosarcomas (n=2). The adult-spindle-cell type was separately categorized because it is considered distinct from pediatric homonymous tumor (ie, a subset of the embryonal type) because of its more aggressive behavior and frequent association with sclerosing features.7
ALK Immunohistochemistry
Four-micrometer-thick sections of the tumors were deparaffinized. Heat-induced epitope retrieval was performed using the targeted retrieval solution pH9 (Dako, Carpinteria, CA, USA). The slides were treated with 3% hydrogen peroxide for 20 min to block endogenous peroxidase activity. They were then incubated with primary antibody against ALK protein (1:40, 5A4; Abcam, Cambridge, UK) for 30 min at room temperature. Immunoreactions were detected using EnVision-FLEX+ (Dako). The reactions were visualized using 3,3′ diaminobenzidine followed by counterstaining with hematoxylin. We had previously shown that this staining condition yielded perfect concordance between immunoreactivity and the ALK rearrangement status in lung cancers.24 The staining results were categorized as negative (<5%), focally positive (5—50%), and diffusely positive (>50%), whereas the staining intensity was graded as weak, moderate, or strong.
Correlation with Clinical Parameters
The clinical data were correlated with the ALK immunoexpression status in the case of alveolar rhabdomyosarcoma. The clinical parameters studied were age, sex, primary tumor site (head and neck, urogenital/perianal, extremity, or trunk), whether the tumor initially presented with metastatic spread to the lymph nodes or distant sites, whether the tested specimen had been obtained before or after chemotherapy, whether the tested specimen was sampled from the primary site or metastatic site, and overall survival.
Correlation with Histological Parameters
Select histological parameters were correlated with the ALK immunoexpression status in the case of alveolar rhabdomyosarcoma. The histological parameters studied were the presence of a pure solid growth pattern, the presence of a mixture with additional embryonal rhabdomyosarcomatous morphology, the presence of nuclear pleomorphism (at least at a focal level), and the percentage of myogenin-immunopositive cells.
FOXO1 and ALK Gene Rearrangement Status
Fluorescence in situ hybridization (FISH) analysis to detect gene rearrangement was performed on formalin-fixed, paraffin-embedded, 4-μm-thick tumor sections. Break-apart probes were used for the FOXO1 (Vysis LSI FOXO1 Dual Color, Abbott Molecular, Abbott Park, IL, USA) and ALK genes (Vysis LSI ALK Dual Color, Abbott Molecular) in accordance with the manufacturer’s instructions. At least 50 nonoverlapping tumor cells were examined. Cases in which >20% of the cells showed split signals or isolated 3′ signals were considered positive for gene rearrangement.
ALK Copy Number Evaluation
Changes in the ALK copy number were determined using FISH on formalin-fixed, paraffin-embedded 4-μm-thick tumor sections. A dual-color probe set was used, in which the ALK gene was labeled with Texas Red (red) and the paracentromeric sequence of the chromosome 2 (CEN2p) was labeled with FITC (green) (GSP laboratory, Kawasaki, Kanagawa, Japan). FISH images were captured using the Metafer Slide Scanning Platform (MetaSystems, Altlussheim, Germany) to facilitate analysis. In order to minimize the influence of truncation artifacts, we evaluated 50−100 nonoverlapping tumor cells with an adequate nuclear area that exhibited at least one each of green and red signals. The specific ALK gene copy number was analyzed by determining the ratio of the copy number of ALK to that of CEN2p, and copy number gain was classified as no selective gain (median ALK/CEN2p=1), low-level selective gain (1<median ALK/CEN2p<2), or selective amplification (median ALK/CEN2p≥2). When there was no selective copy number gain (ie, median ALK/CEN2p=1), we focused on the absolute ALK copy number per cell. An increase in this parameter was categorized as no polysomy (≥4 ALK gene copy in <10% of cells), low polysomy (≥4 ALK gene copy in 10−<40% of cells), and high polysomy (≥4 ALK gene copy in ≥40% cells), similar to the criteria widely used in thoracic oncology.26
ALK Mutation Analysis
We extracted DNA from formalin-fixed paraffin-embedded tumor sections. Exons 23 and 25 of the ALK gene, which harbor hotspots of activating mutations, were amplified by PCR using EX Taq HS polymerase (TAKARA, Otsu, Shiga, Japan). The primers used were ALK-EX23F, 5′-GCCTTTATACATTGTAGCTGC-3′; ALK-EX23R, 5′-TCGGAGGAAGGACTTGAGGTC-3′; ALK-EX25F, 5′-TCTTCCCAGAGACATTGCTGC-3′; and ALK-EX25R, 5′-GGTAGAAAGTTGACAGGGTAC-3′ The PCR products were purified (QIAquick PCR purification kit; Qiagen, Valencia, CA, USA) and analyzed by sequencing with the same primers (Big Dye sequencing kit; Applied Biosystems, Carlsbad, CA, USA). Somatic mutations were evaluated by sequencing matched normal DNA extracted from muscle tissues. Somatic amino acid substitution was functionally evaluated using the Polyphen2 and MutationAssessor algorithms (http://genetics.bwh.harvard.edu/pph2/ and http://mutationassessor.org/, respectively).
Statistical Analysis
All data analyses were performed using SPSS version 20.0 (IBM Corporation, Somers, NY, USA). Fisher’s exact test was used for categorical data, and Mann–Whitney’s U test was used for continuous data. Overall survival, measured from the date of clinical presentation, was calculated using the Kaplan–Meier method, and survival difference was compared using the log-rank test. All P values were two tailed, and P<0.05 was considered significant.
Results
Correlation between ALK Expression and Tumor Subtype
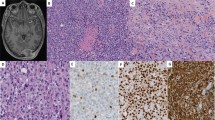
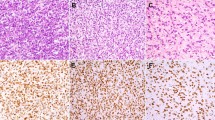
As shown in Table 1, ALK expression was detected in 2 (6%) of 33 embryonal, 42 (69%) of 61 alveolar, 0 (0%) of 8 pleomorphic, 0 (0%) of 12 adult-spindle-cell/sclerosing, and 0 (0%) of 2 epithelioid rhabdomyosarcomas. ALK expression was significantly associated with tumor subtype (P<0.001). Among the 44 ALK-positive cases, 11 showed strong staining intensity, 14 showed moderate intensity, and 19 showed weak intensity (Figure 1). The staining extent was diffuse in 40 cases and focal in 4 cases. The median rate of positive cells was 80%. In immunopositive cases, the reaction was at the plasma membrane and/or cytoplasm with frequent paranuclear Golgi accentuation; no nuclear reactivity was seen.

ALK immunoreactivity in rhabdomyosarcomas. The staining was (a) strongly positive, (b) moderately positive, (c) weakly positive, or (d) negative.
Correlation between ALK Expression and Clinicopathologic Parameters in Alveolar Rhabdomyosarcoma
As shown in Table 2, the age, sex, primary tumor site, chemotherapy status, and sample type were not significantly associated with ALK immunoreactivity. ALK-positive and ALK-negative alveolar rhabdomyosarcomas initially presented with metastasis in 67% and 32% of the cases, respectively, and the difference was statistically significant (P=0.014). Further, overall survival did not differ between the groups (P=0.96, Figure 2). Among the histological parameters, pure solid growth pattern and mixture with embryonal morphology were not significantly associated with ALK status. All three alveolar rhabdomyosarcomas with at least focal nuclear pleomorphism were ALK immunonegative (P=0.027). The median rate of myogenin-immunopositive cells in the ALK-positive alveolar rhabdomyosarcomas was 95%, which was significantly higher than that (60%) in ALK-negative ones (P=0.001).

Kaplan–Meier analysis of overall survival of 42 patients with ALK-positive alveolar rhabdomyosarcoma and 19 patients with ALK-negative alveolar rhabdomyosarcoma. No significant difference in survival was observed between the groups as determined by the log-rank test (P=0.96).
Correlation between ALK Expression and FOXO1 Status in Alveolar rhabdomyosarcoma
FOXO1 gene rearrangement status was examined in all the alveolar rhabdomyosarcomas and select non-alveolar cases. Among 47 alveolar tumors successfully studied, 41 cases (87%) showed FOXO1 rearrangement, while 6 cases were negative for FOXO1 rearrangement. In contrast, none of the 18 non-alveolar tumors studied, including both ALK-positive embryonal cases, showed FOXO1 rearrangement. All the 33 ALK-positive alveolar tumors and 8 of the 14 ALK-negative alveolar tumors showed FOXO1 rearrangement (Table 3). Thus, ALK immunoexpression and FOXO1 status were significantly correlated (P<0.001).
ALK Rearrangement Status
The ALK gene rearrangement status was determined in select ALK-positive and ALK-negative cases. None of the 24 cases (12 ALK positive, 12 ALK negative) tested showed ALK rearrangement.
Changes in ALK Copy Number
Changes in the ALK copy number were examined in all the alveolar rhabdomyosarcomas and select non-alveolar cases. Both ALK-positive embryonal rhabdomyosarcomas were included in the analysis. FISH was successful for 47 alveolar (35 ALK positive and 12 ALK negative) and 13 non-alveolar (2 ALK positive and 11 ALK negative) tumors. The median ALK copy number per cell was significantly greater in the ALK-positive tumors than the ALK-negative ones (P=0.005, Figure 3). However, the median ALK/CEN2p ratio did not differ significantly between these tumors (P=0.106, Figure 4), and most cases (56 cases, 93%) showed no selective increase in the ALK gene dosage (median ALK/CEN2p=1, Figure 5a). Selective amplification was seen in one alveolar rhabdomyosarcoma (median ALK/CEN2p=7, Figure 5b), which was that of a post-chemotherapy metastatic tumor that primarily occurred in the finger of an adult man, and the tumor showed strong diffuse ALK expression. Low selective gain was identified in one alveolar and two embryonal tumors (Figure 5c), and all three cases were positive for ALK expression. Tumor cells with ≥4 ALK copies were significantly more common in ALK-positive tumors than in ALK-negative tumors (P=0.013, Figure 6). Among the 56 cases that showed no selective increase in the ALK copy number, 29 showed no polysomy, 20 showed low polysomy, and 7 showed high polysomy (Figure 5d). The latter pattern did not seem to merely represent the near-tetraploidy typical of alveolar rhabdomyosarcoma,27, 28 because six of the seven tumors with this pattern consisted of ≥10% cells with ≥5 ALK copies. All seven cases with the high-polysomy pattern were ALK immunopositive, while 26 ALK-positive and 23 ALK-negative tumors showed the no- or low-polysomy pattern. In summary, all 11 cases with selective ALK amplification, low selective gain, or high polysomy were ALK immunopositive. Overall, these genomic abnormalities were uncommon, accounting for 18% of the 60 rhabdomyosarcomas tested and 30% of the 37 ALK-positive rhabdomyosarcomas.

The median ALK copy number per cell determined by FISH in 37 ALK-positive and 23 ALK-negative rhabdomyosarcomas. ▴ indicates the alveolar subtype, and ○ indicates non-alveolar subtypes. ALK-positive tumors carried a significantly greater number of ALK gene copies than ALK-negative tumors did (P=0.005).

The median ALK/CEN2p ratio determined by FISH in 37 ALK-positive and 23 ALK-negative rhabdomyosarcomas. ▴ indicates the alveolar subtype, and ○ indicates non-alveolar subtypes. The vast majority (93%) of tumors showed a median ALK/CEN2p ratio of 1, and collectively, the ratio was not significantly different between the ALK-positive and ALK-negative groups (P=0.106). However, all four cases with a median ALK/CEN2p >1 were ALK immunopositive.

Changes in the ALK copy number in rhabdomyosarcomas determined by dual-color FISH. Red signals represent the ALK gene and green signals represent the reference sequence (CEN2p). (a) Most tumors tested showed no selective increase in ALK gene dosage (ALK/CEN2p=1). (b) One tumor showed selective ALK amplification. (c) Three tumors showed low selective gain defined as 1<median ALK/CEN2p<2. (d) Seven tumors showed a high-polysomy pattern defined as ≥4 ALK copies in ≥40% cells with ALK/CEN2p=1.

The percentage of tumor cells that harbored ≥4 copies of ALK gene determined by FISH in 37 ALK-positive and 23 ALK-negative rhabdomyosarcomas. ▴ indicates the alveolar subtype, and ○ indicates non-alveolar subtypes. ALK-positive tumors carried a significantly greater percentage of cells with ≥4 copies of the ALK gene than ALK-negative tumors did (P=0.013).
ALK Mutation Status
ALK mutation was examined in 34 cases, and the analysis was successful in 19 cases (7 ALK positive, 12 ALK negative). Only one case, a metastatic embryonal rhabdomyosarcoma in an adult male, showed a heterozygous somatic mutation (c.3830G>A, p.I1277T) of the receptor tyrosine kinase domain. This tumor was immunonegative for ALK and was located primarily in the heart. Functional evaluation of amino acid substitution using Polyphen2 and MutationAssessor predicted that this mutation may cause harmful effects (probably damaging or medium functional impact). This substitution is not found in the Catalog of Somatic Mutations in Cancer (http://www.sanger.ac.uk/genetics/CGP/cosmic/).
Discussion
Previous studies showed that alveolar rhabdomyosarcomas were more commonly immunopositive for ALK than other subtypes were (Table 4). Our series showed an even sharper difference in ALK reactivity between alveolar (69%) and other (4%) subtypes, which may be because of the differences in the staining protocol. Specifically, we used the 5A4 antibody and a polymer-based system, whereas most prior studies applied a more conventional method using the ALK1 antibody and the avidin-biotin system. Recent reports have suggested that ALK1/avidin-biotin method may result in suboptimal concordance between immunoreactivity and genomic status.29, 30, 31 The differential ALK staining in rhabdomyosarcoma may carry diagnostic value. Subtyping rhabdomyosarcoma can be challenging, particularly when alveolar tumor shows a solid pattern and embryonal tumor shows dense cellularity without an apparent myxoid matrix.32 In addition, sclerosing rhabdomyosarcoma may mimic the alveolar subtype.4, 6 Although FOXO1 rearrangement assays may be useful for classification, immunohistochemical analysis is more accessible and cost effective. ALK staining has already gained wide application in diagnostic pathology, and optimized staining protocols like the one used here may become prevalent given the increased interest in ALK-targeted therapy. Our data suggest that ALK may be a useful marker for distinguishing the alveolar subtype from other subtypes of rhabdomyosarcomas, and this finding needs to be confirmed in a larger cohort.
ALK-positive and ALK-negative alveolar rhabdomyosarcomas were not clinicopathologically different in many respects, including overall survival. However, we found that ALK-positive cases presented with metastasis more commonly than ALK-negative cases. Although additional studies are needed for confirmation, it appears that ALK immunoreactivity may identify a subset of alveolar rhabdomyosarcoma that has a higher proclivity to metastatic spread. On histological examination, nuclear pleomorphism was exclusively observed in ALK-negative alveolar rhabdomyosarcomas, and ALK-negative alveolar cases harbored a smaller proportion of myogenin-positive cells than ALK-positive ones. Classic alveolar rhabdomyosarcoma is characterized by uniform cytology and a greater extent of myogenin staining compared with non-alveolar subtype.25, 33 Therefore, the presence of pleomorphic tumors and less extensive myogenin positivity in the ALK-negative alveolar cases may suggest that this subgroup includes non-classical examples. Some of these might be close to (or might actually represent) the non-alveolar subtype that is indistinguishable from alveolar rhabdomyosarcoma on the basis of morphological criteria alone.
ALK immunoreactivity and FOXO1 status were strongly correlated in our cohort. This result is in agreement with several array-based analyses34, 35, 36, 37 in which ALK was found to be one of the most differentially expressed genes in FOXO1 fusion-positive alveolar rhabdomyosarcoma as compared with fusion-negative alveolar and embryonal rhabdomyosarcoma. This association indicates that ALK may be a target of the PAX-FOXO1 chimeric protein. In support of this hypothesis, Davicioni et al36 showed that ALK was upregulated when PAX-FOXO1 was transfected into a fusion-negative cell line. Cao and associates38 also showed that introducing small hairpin RNA against PAX3-FOXO1 into a fusion-positive cell line downregulated ALK expression. Further, the regulatory sequences of the ALK gene were shown to contain a high-affinity binding site to the PAX3-FOXO1 protein.38 Although a previous study20 found no association between FOXO1 rearrangement and ALK immunoreactivity, this discordance may be owing to the difference in the number of cases studied, the ALK staining protocol used, and the molecular method employed to determine the gene rearrangement status.
The ALK immunoreactivity and ALK gene copy number were also found to be significantly correlated in our series. Notably, ALK selective amplification, low selective gain, and high polysomy were observed exclusively in ALK-immunopositive cases. Nevertheless, these significant copy number changes were relatively uncommon and were observed only in 30% of the ALK-positive tumors. In particular, true selective gene amplification was rare: it was observed in only 3% of the ALK-positive cases. This finding is consistent with that of a previous report in which rhabdomyosarcoma cell lines were found not to harbor DNA copy number gain at the 2p23 locus, where the ALK gene is located.39 Similarly, ALK was not found to be a differentially amplified gene when genomic imbalances were compared between primary rhabdomyosarcoma tissues and normal muscle.39
Our finding of a low incidence of increased ALK copy number in rhabdomyosarcoma was in contrast with the results of a recent large study by van Gaal et al23 In their study, the absolute ALK copy number increased (>4 copies in >10% of cells) in 88% of alveolar rhabdomyosarcomas and 52% of embryonal tumors, and a selective ALK gain (average ALK/LAF>1.5) was seen in 45% of alveolar tumors and 61% of embryonal cases. The reason for this incongruence is unclear. Even when FISH interpretation criteria similar to theirs were applied, the ALK gain/amplification was uncommon in our cohort: tumors with >4 ALK copies in >10% of cells were observed in 23% of the alveolar tumors and 23% of the non-alveolar subtypes, and an average ALK/CEN2p of >1.5 was seen in 4% of the alveolar tumors and 15% of the non-alveolar cases. Other investigators also reported ALK gene gain/amplification in rhabdomyosarcoma (Table 4), but the number of cases studied was small, and the quantitative definition of gain/amplification was not provided.19, 20, 22 Notably, these prior studies either did not use a reference probe19, 22 or separately examined the ALK gene and a reference gene,20, 23 whereas we simultaneously evaluated the ALK and reference genes using a dual-color approach that enabled precise determination of gene copy number at the cellular level.
ALK gene rearrangement and somatic mutation do not seem to have a major role in rhabdomyosarcoma. ALK rearrangement was not identified in any of the cases examined in this study, a finding similar to that of other studies.20, 22, 23 Interestingly, one study19 found ALK rearrangement in three of six rhabdomyosarcomas tested. We identified an ALK missense mutation (c.3830G>A, p.I1277T) in one embryonal rhabdomyosarcoma. However, this tumor was not immunopositive for ALK, indicating that the mutation may not be an activating one. This mutation was different from that (c.3673G>A, p.D1225N) reported previously in an embryonal rhabdomyosarcoma.23 The same study23 also identified frame-shift deletions in two alveolar and five embryonal rhabdomyosarcomas by cDNA sequencing, which probably represented alternative mRNA splicing;23 we did not find any deletions in the genomic DNA.
The positive ALK immunoreactivity in a subset of rhabdomyosarcomas, some of which are associated with an ALK gene copy number increase, may suggest the potential clinical benefit of ALK inhibition. However, whether ALK expression without gene rearrangement has oncologic relevance remains controversial. Thus far, ALK-targeted therapy has been shown to be clinically effective only in ALK-rearranged tumors. Some researchers40 argued that ALK expression in rhabdomyosarcoma may simply be physiological rather than oncogenic, by showing that the ALK-expressing Rh30 cell line lacked ALK autophosphorylation. In contrast, others41, 42, 43 suggested that in neuroblastomas, ALK protein expression itself exerts oncogenic activity irrespective of genomic status and that it is a potential target of kinase inhibitors. Future clinical studies should investigate whether ALK inhibitors are beneficial for treating ALK-immunopositive tumors without gene rearrangement.
In summary, we clarified the subtype-specific incidence of ALK immunoreactivity in rhabdomyosarcomas and showed the association between ALK expression and several clinicopathologic/molecular parameters. This comprehensive analysis will serve as a basis for future investigations that aim to determine the indication, or lack thereof, of ALK-targeted therapy for patients with rhabdomyosarcoma.
References
Montgomery E, Barr FG . Pleomorphic rhabdomyosarcoma In: Fletcher CDM, Unni KK, Mertens F, (eds) Pathology and genetics of tumours of soft tissue and bone. IARC Press: Lyon, France, 2002, pp 153–154.
Parham D, Barr FG . Embryonal rhabdomyosarcoma In: Fletcher CDM, Unni KK, Mertens F, (eds) Pathology and genetics of tumours of soft tissue and bone. IARC Press: Lyon, France, 2002, pp 146–149.
Parham D, Barr FG . Alveolar rhabdomyosarcoma In: Fletcher CDM, Unni KK, Mertens F, (eds) Pathology and genetics of tumours of soft tissue and bone. IARC Press: Lyon, France, 2002, pp 150–152.
Folpe AL, McKenney JK, Bridge JA et al. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol 2002;26:1175–1183.
Jo VY, Marino-Enriquez A, Fletcher CD . Epithelioid rhabdomyosarcoma: clinicopathologic analysis of 16 cases of a morphologically distinct variant of rhabdomyosarcoma. Am J Surg Pathol 2011;35:1523–1530.
Mentzel T, Katenkamp D . Sclerosing pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch 2000;436:305–311.
Nascimento AF, Fletcher CD . Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol 2005;29:1106–1113.
Morris SW, Kirstein MN, Valentine MB et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994;263:1281–1284.
Gascoyne RD, Lamant L, Martin-Subero JI et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood 2003;102:2568–2573.
Lawrence B, Perez-Atayde A, Hibbard MK et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377–384.
Soda M, Choi YL, Enomoto M et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–566.
Debelenko LV, Raimondi SC, Daw N et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol 2011;24:430–442.
Marino-Enriquez A, Ou WB, Weldon CB et al. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer 2011;50:146–153.
Chen Y, Takita J, Choi YL et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008;455:971–974.
Murugan AK, Xing M . Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res 2011;71:4403–4411.
Kwak EL, Bang YJ, Camidge DR et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693–1703.
Butrynski JE, D'Adamo DR, Hornick JL et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 2010;363:1727–1733.
Gambacorti-Passerini C, Messa C, Pogliani EM . Crizotinib in anaplastic large-cell lymphoma. N Engl J Med 2011;364:775–776.
Cessna MH, Zhou H, Sanger WG et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol 2002;15:931–938.
Corao DA, Biegel JA, Coffin CM et al. ALK expression in rhabdomyosarcomas: correlation with histologic subtype and fusion status. Pediatr Dev Pathol 2009;12:275–283.
Pillay K, Govender D, Chetty R . ALK protein expression in rhabdomyosarcomas. Histopathology 2002;41:461–467.
Li XQ, Hisaoka M, Shi DR et al. Expression of anaplastic lymphoma kinase in soft tissue tumors: an immunohistochemical and molecular study of 249 cases. Hum Pathol 2004;35:711–721.
van Gaal JC, Flucke UE, Roeffen MH et al. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J Clin Oncol 2012;30:308–315.
Yoshida A, Tsuta K, Nakamura H et al. Comprehensive histologic analysis of ALK-rearranged lung carcinomas. Am J Surg Pathol 2011;35:1226–1234.
Morotti RA, Nicol KK, Parham DM et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children's Oncology Group experience. Am J Surg Pathol 2006;30:962–968.
Cappuzzo F, Hirsch FR, Rossi E et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 2005;97:643–655.
Kilpatrick SE, Teot LA, Geisinger KR et al. Relationship of DNA ploidy to histology and prognosis in rhabdomyosarcoma. Comparison of flow cytometry and image analysis. Cancer 1994;74:3227–3233.
Shapiro DN, Parham DM, Douglass EC et al. Relationship of tumor-cell ploidy to histologic subtype and treatment outcome in children and adolescents with unresectable rhabdomyosarcoma. J Clin Oncol 1991;9:159–166.
Mino-Kenudson M, Chirieac LR, Law K et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res 2010;16:1561–1571.
Takeuchi K, Choi YL, Togashi Y et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res 2009;15:3143–3149.
Takeuchi K, Soda M, Togashi Y et al. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin Cancer Res 2011;17:3341–3348.
Parham DM, Qualman SJ, Teot L et al. Correlation between histology and PAX/FKHR fusion status in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Am J Surg Pathol 2007;31:895–901.
Dias P, Chen B, Dilday B et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol 2000;156:399–408.
Williamson D, Missiaglia E, de Reynies A et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 2010;28:2151–2158.
Wachtel M, Dettling M, Koscielniak E et al. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res 2004;64:5539–5545.
Davicioni E, Finckenstein FG, Shahbazian V et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 2006;66:6936–6946.
Lae M, Ahn EH, Mercado GE et al. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J Pathol 2007;212:143–151.
Cao L, Yu Y, Bilke S et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res 2010;70:6497–6508.
Missiaglia E, Selfe J, Hamdi M et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer 2009;48:455–467.
Lamant L, Pulford K, Bischof D et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol 2000;156:1711–1721.
Passoni L, Longo L, Collini P et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res 2009;69:7338–7346.
De Brouwer S, De Preter K, Kumps C et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res 2010;16:4353–4362.
Duijkers FA, Gaal J, Meijerink JP et al. Anaplastic lymphoma kinase (ALK) inhibitor response in neuroblastoma is highly correlated with ALK mutation status, ALK mRNA and protein levels. Cell Oncol 2011;34:409–417.
Acknowledgements
We thank Dr David Parham for helpful comments, and Sachiko Miura and Chizu Kina for superb technical assistance. This work was supported in part by the National Cancer Center Research and Development Fund (23-A-10). Presented in part at the 101st Annual Meeting of the United States and Canadian Academy of Pathology in Vancouver, BC, Canada, March 2012.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yoshida, A., Shibata, T., Wakai, S. et al. Anaplastic lymphoma kinase status in rhabdomyosarcomas. Mod Pathol 26, 772–781 (2013). https://doi.org/10.1038/modpathol.2012.222
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.222
Keywords
This article is cited by
-
Synchronous lung and multiple soft tissue metastases developed from osteosarcoma of tibia: a rare case report and genetic profile analysis
BMC Musculoskeletal Disorders (2022)
-
A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation
Modern Pathology (2020)
-
OLIG2 is a novel immunohistochemical marker associated with the presence of PAX3/7-FOXO1 translocation in rhabdomyosarcomas
Diagnostic Pathology (2019)
-
Targeting ALK in pediatric RMS does not induce antitumor activity in vivo
Cancer Chemotherapy and Pharmacology (2018)
-
Targeting Anaplastic Lymphoma Kinase (ALK) in Rhabdomyosarcoma (RMS) with the Second-Generation ALK Inhibitor Ceritinib
Targeted Oncology (2017)
