
Abstract
Metastasis is the main cause of mortality in patients with colorectal cancer. However, most of the targeted therapies and predictive molecular biomarkers were developed based mainly on primary tumors. The current study was conducted to determine the degree of discordance between potential predictive and/or prognostic molecular markers in primary colorectal tumors and corresponding metastases, as this could have an impact on the efficacy of targeted therapies in the advanced colorectal cancer. KRAS, PIK3CA and BRAF mutations were determined by Sanger sequencing and mutant-enriched polymerase chain reaction (PCR) assays in 83 paired samples, MET gene copy number by quantitative PCR in 59, MET expression by immunohistochemistry in 73 and nuclear and cytoplasmic expression of PTEN by immunohistochemistry in 78 and 71 pairs, respectively. A certain degree of discordance between primary tumors and corresponding metastases was demonstrated for all examined biomarkers except BRAF mutations. PIK3CA exon 9 mutations in primary tumors and loss of PTEN nuclear expression in metastases correlated with KRAS mutations. KRAS wild-type status in primary tumors was associated with loss of PTEN cytoplasmic expression and high gene copy number of MET. Survival and clinical data were available for 68 patients. The multiple regression analysis revealed that the right-sided tumor localization and overexpression of MET were associated with shorter overall survival.
Similar content being viewed by others

Main
Colorectal cancer is the third more common tumor and the second reason of cancer-related deaths worldwide. About 25% of patients with colorectal cancer present metastases at the time of diagnosis and ∼50% of patients who undergo radical resection for primary colorectal cancer present disease dissemination during the first 5 years from diagnosis. Treatment options for patients with advanced disease have been expanded combining anti-EGFR or anti-VEGF monoclonal antibodies with chemotherapy.1, 2
The administration of anti-EGFR monoclonal antibodies has been associated with antitumor activity in 10–20% of patients with metastatic colorectal cancer; however, recent studies suggested that the anti-EGFR-mediated antitumor activity is restricted to patients with wild-type KRAS tumors. Conversely, up to 65% of KRAS wild-type patients do not benefit from the EGFR-targeted therapy and mutations in other downstream effectors of the EGFR signaling pathway such as BRAF, NRAS, PIK3CA and loss of expression of PTEN seem to be responsible for this phenomenon.3, 4, 5, 6 However, the negative selection of the four mutant genotypes (KRAS, BRAF, NRAS, PIK3CA) offers modest improvements in terms of objective response rates compared with KRAS, indicating that additional markers are needed in order to better predict response to EGFR-targeted therapy.3 Therefore, taking into account the cross talk between the EGFR and MET signaling pathways, the activation of the MET receptor tyrosine kinase pathway has also been investigated as another possible mechanism of resistance to EGFR-targeted therapy.7, 8
The majority of studies aiming to identify effective predictive molecular markers have been conducted using the tissue obtained from primary tumors. This has been mainly based on the hypothesis that metastases were fundamentally similar to primary tumors. Indeed, it has been shown that most of mutations present in metastases could also be detected in primary tumors, suggesting that migrating and metastases-generating cells already exist among the initial cell clones present in the primary tumor.9, 10 Consistently, KRAS and BRAF mutation status were concordant between primary tumors and corresponding distant metastases in ∼90–100% of the cases while only few reports exist on the PIK3CA mutation status.11, 12, 13 Despite these data, recent studies have demonstrated the presence of genotypic differences between the primary tumors and the metastatic lesions.14 The genetic analyses of disseminated tumor cells and the identification of gene expression profiles that predict the metastatic potential suggested that metastasis is predetermined early during tumorigenesis.15, 16 Moreover, several reports have shown that more genes were methylated in primary colorectal tumors than in corresponding metastatic lesions and metastases can exhibit substantial differences in gene expression patterns compared with primary tumors.17, 18, 19
In view of these data, the heterogeneity between primary tumors and metastases emerged as an additional reason for the failure of targeted therapies in colorectal cancer; thus, the current study was conducted in order to determine the degree of discordance between potential predictive and/or prognostic molecular markers in primary tumors and corresponding metastases, and to investigate the possible associations between these biomarkers in primary tumors, as well as in metastases.
Materials and methods
Patients
The archive of the Department of Pathology of the University Hospital of Heraklion, Greece, was searched retrospectively for patients who had matched samples of colorectal primary tumors and synchronous or metachronous metastases. The search period spanned from 1998 to 2010. The samples were fixed in neutral-buffered formalin and embedded in paraffin. The hematoxylin–eosin (HE) sections from the colorectal primary tumor and metastases specimens were reviewed by a pathologist expert in gastrointestinal cancer to confirm the presence of tumor tissue.
All patients had undergone either oncologic resection or tissue biopsy of the tumors for diagnostic and therapeutic purposes.
Tumors occurring at or proximal to the splenic flexure were considered right-sided (proximal) and those in the descending and sigmoid colon were left-sided (distal). Synchronous metastases were defined as metastases detected by preoperative imaging on computed tomography or magnetic resonance imaging or during resection of the primary tumor. Metachronous metastases were detected during follow-up. Clinical data for eligible patients were obtained from the review of all available medical records regarding hospitalization and outpatient visits at the Oncology Department of the University Hospital of Heraklion. Baseline performance status was determined by the treating physician according to Eastern Cooperative Oncology Group criteria. For cancer-specific survival analysis, death attributed to colon cancer was defined as a clinical end point. Overall survival was defined as the interval between diagnosis to death or last follow-up visit.
Mutational Analysis
Tumor tissues to be genotyped were marked on standard HE-stained histologic slides; in order to achieve at least 80% tumor tissue for DNA extraction, 4 μm serial sections were manually macrodissected or microdissected using the Eppendorf Piezo-Power Microdissection system (Eppendorf, Hamburg, Germany). Genomic DNA was extracted using the QIAamp DNA Micro kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Mutational analyses of KRAS by Sanger sequencing were performed as previously described.20 Exons 9 and 20 of the PIK3CA were sequentially amplified by two rounds of polymerase chain reaction (PCR) and subjected to by-directional automatic sequencing. The PCR primers for PIK3CA amplification were as follows: 99U(exon 9F)- 5′-ACAACAGTTAATTAGCAATGTAAAA-3′; 220U(exon 9F)- 5′-ACAGAGTAACAGACTAGCTAGAGAC-3′; 408L(exon 9R)- 5′-ACATGCTGAGATCAGCCAAAT-3′; 37U(exon 20F)- 5′-TCGACAGCATGCCAATCTC-3′; 45U(exon 20F)- 5′-ATGCCAATCTCTTCATAAATC-3′; 283L(exon 20R)- 5′-CAGAGTGAGCTTTCATTTTCT-3′. Sanger sequencing has a sensitivity of ∼20%.
KRAS TaqMan-MGB allelic discrimination assays were designed (custom TaqMan® SNP Genotyping Assays, Applied Biosystems, Foster City, CA, USA) to detect the seven more common KRAS mutations; (c.35G>A (p.G12D), c.38G>A (p.G13D), c.34G>C (p.G12R), c.34G>T (p.G12C), c.34G>A (p.G12S), c.35G>T (p.G12V) and c.35G>C (p.G12A)). The amplicons were set at 80 bp. The reporter fluorophores for TaqMan-MGB probes were VIC for wild type and FAM for each KRAS-mutant allele. The PCR primers and probes are available upon request. The peptide nucleic acid (PNA) clamp designed to hybridize to the wild-type KRAS allele surrounding codons 12 and 13 was as follows: H2N-CCTACGCCACCAGCTCC-CO-N2H (Eurogentec S.A., Seraing, Belgium). The PNA/DNA hybrid is unstable due to base pair mismatches; therefore, PNA hybridization inhibits the annealing of the wild-type KRAS TaqMan-MGB probes but does not inhibit the annealing of the KRAS-mutant TaqMan-MGB probes. The reaction was performed in a total volume of 10 μl, containing 2 μl extracted DNA (20 ng), 5 μl 2 × TaqMan® genotyping master mix (Applied Biosystems), 0.25 μl of genotyping assay mix and 200 nM PNA; in parallel, a non-PNA-clamp reaction was performed without any PNAs. The PCR conditions were 95 °C for 10 min, followed by two-step cycling: 50 cycles of 92 °C for 15 s, and 62 °C for 90 s. In each experiment, PCR reagents without template were run in parallel as the no-template control. The sample experiment data and results were generated using an ABI 7900 real-time PCR system (Applied Biosystems) by performing an amplification run using a standard curve plate document to generate real-time PCR data and an allelic discrimination run using an allelic discrimination plate document. The fluorescence data and amplification plots were analyzed with the SDS 2.3 software (Applied Biosystems). The evaluation of KRAS mutation status was based on the cycle thresholds of the wild-type and mutant amplification curves as compared between the PNA and non-PNA reactions.
The assay analytical specificity was evaluated using genomic DNA (0.2, 20 ng) isolated from cell lines wild-type (HT-29) or positive (LS174, c.35G>A (p.G12D); HCT116, c.38G>A (p.G13D); HUP-T3, c.34G>C (p.G12R); KYSE410, c.34G>T (p.G12C); A549, c.34G>A (p.G12S); SW403, c.35G>T (p.G12V) and RPMI8226, c.35G>C (p.G12A)) for specific KRAS mutations and confirmed by sequencing. All cell lines were obtained from the A. Jung laboratory (Department of Pathology, Ludwig-Maximilians Universitat Munchen, Germany) and were cultured according to the supplier’s recommendations. To assess analytical sensitivity, each of the seven KRAS-positive cell line genomic DNAs was serially diluted in a background of KRAS wild-type genomic DNA provided by the HT-29 cell line to give mutation/wild-type ratios of 100, 50, 20, 10, 5, 1, 0.2 and 0%. For example, the 1:100 mixtures contained 1 ng of mutant DNA and 100 ng of wild-type DNA. Analysis of the mean signal generated by the positive mutant probes plus PNA showed that the assay can reproducibly detect KRAS mutations in 0.2% genomic DNA dilution.
BRAF c.1799T>A (p.V600E) mutation detection was assessed by allelic discrimination using Taqman-MGB probes (custom TaqMan® SNP Genotyping Assays, Applied Biosystems). The amplification was performed on an ABI 7900 real-time PCR system (Applied Biosystems) in an 10 μl mixture composed of 2 μl of extracted DNA (20 ng), 5 μl of 2 × TaqMan® genotyping master mix (Applied Biosystems) and 0.25 μl of genotyping assay mix containing primers and probes (available on request). The reaction was performed with a preliminary step for 2 min at 50 °C, followed by denaturation for 10 min at 95 °C, then 50 cycles of two steps at 92 °C for 15 s and 62 °C for 90 s. Allelic discrimination was performed using SDS 2.3 software (Applied Biosystems).
ARMS allele-specific PCR combined with Scorpions probes for the detection of PIK3CA mutations (c.1633G>A (p.E545K), c.1624G>A (p.E542K), c.3140A>G (p.H1047R)) was performed as previously described.21
Quantitative Real-Time PCR
Relative MET amplification levels were determined by quantitative real-time PCR by comparison with the amplification levels of TOP3A residing at chromosome locus 17p11 using the following primers: MET-sense: 5'-AATTGTGTCTTTCTCTAGGCATGTCA-3'; MET-antisense: 5'-GGGAAGGAGTGGTACAACAGATTATC-3', MET probe FAM-CATCGCTCTAATTCAG-NFQ, TOP3A-sense: 5'-CCACTGCGAACTTAAGAAAACTTTG-3'; TOP3A-antisense: 5'-TTCTCTATCACAGTCAGTCCAGATCA-3', TOP3A probe VIC-AACGAGAGACTCGCCAGT-NFQ.22 Genomic DNA from formalin-fixed paraffin embedded tissue samples, positive control (H1993 cell line for MET amplification), and reference (pooled PBMCs from healthy volunteers) (20 ng) was amplified for 40 cycles (15 s, 95 °C; 60 s, 60 °C) in a ABI 7900 real-time PCR system (Applied Biosystems), using the Platinum® qPCR SuperMix-UDG (Invitrogen, Carlsbad, CA, USA), 900 nM primers and 150 mM probes. Sensitivity of the assay was assessed by diluting H1993 cells with PBMCs from healthy volunteers or H2073 cells, which do not harbor MET amplification. Amplification of MET was clearly distinguished in DNA samples from 10% of H1993 cells mixed with 90% PBMCs (or 90% H2073) from those of PBMCs (or H2073 cells). Efficiency of the PCR reaction in each assay was assessed by a four point standard curve (125, 12.5, 1.25 and 0.125 ng) using DNA from pooled PBMCs. As relative amplification levels were considered the fold changes calculated using the equation 2-ΔΔCT, where ΔΔCT=(CT(MET)sample−CT(TOP3A)sample) −(CT(MET)reference DNA−CT(TOP3A)reference DNA); the reference sample consisted of DNA extracted from PBMCs of healthy volunteers. The cutoff values used to claim MET amplification were established from qPCR data of 14 normal lung formalin-fixed paraffin embedded samples. A tumor sample was considered as amplified when its fold change is over the mean fold change calculated from the 14 normal formalin-fixed paraffin embedded samples+two standard deviations. For all patients, triplicate cycle time (CT) values were averaged.
Immunohistochemistry
Immunostaining was performed on formalin-fixed paraffin wax embedded tissue sections, using UltraVision LP Large Volume Detection System AP Polymer (Thermo Fisher Scientific, Fremont, CA, USA ). Primary antibodies for nuclear PTEN (Ms Mab, Clone 28H6 Catalog No. sc-56205; dilution 1:200, Santa Cruz, Biotechnology, USA), cytoplasmic PTEN (Ms Mab, Clone 17.A, Catalog No. MS1601; dilution 1:25, Neomarkers, Lab Vision, Fremont, CA, USA) and MET (Rb Pab, Clone C-28, Catalog No sc-161; dilution 1:100, Santa Cruz) were used. A step of microwave heating in a solution of sodium citrate was performed before incubation with nuclear PTEN and MET primary antibodies; a step of EDTA antigen retrieval was used for cytoplasmic PTEN. Positive control slide was included for MET consisted of paraffin wax sections from a case of melanoma (membranous or cytoplasmic immunostaining was classified as positive). Paraffin wax sections of prostate cancer and endothelial cells were used as external and internal positive controls, respectively, for cytoplasmic PTEN. Nuclear immunostaining of lymphocytes and stroma cells served as internal positive control for nuclear PTEN. Negative control slides were prepared by omitting the primary antibody.
PTEN cytoplasmic and nuclear expression levels were scored semiquantitatively as previously described.23, 24 Intensity of immunoreactivity for both antibodies was determined as 0, negative; 1, weak; 2, moderate; and 3, strong. As regards the interpretation of PTEN cytoplasmic expression, one, two or three additional points were attributed if the percentage of positive cells was <25%, 25–50% or>50%; tumors with a score <4 were considered to have loss of PTEN cytoplasmic expression. As regards the interpretation of PTEN nuclear expression, tumors with <10% of cancer cells stained with any intensity were considered to have loss of PTEN nuclear expression.
The MET immunoreactivity was predominantly localized in the cytoplasm and membrane, whereas in 5% of tumor specimens a nuclear immunoreactivity could also be detected in cancer cells. MET protein expression levels were arbitrarily scored dependent on the staining intensity as previously described; 0, negative; 1, weak; 2, moderate; and 3, strong.25 Tumors with strong staining intensity were considered as MET overexpressed.
Statistical Analyses
Associations between KRAS, BRAF, PIK3CA mutations, MET gene status, PTEN and MET protein expression and clinicopathologic variables were assessed by χ2-test or Fisher’s exact test. Tested variables included factors related to the patient (age, sex, and performance status) and factors related to the primary tumor (tumor location, histologic type, invasion depth, lymph node status and histological grading). The association between primary tumors and related metastases for KRAS, PIK3CA and BRAF mutational status, MET gene status and PTEN and MET protein expression was evaluated by means of the Cohen’s k-test, appropriate for the assessment of the concordance between two categorical measurements of the same individual. A moderate and good agreement was defined as the coefficient was 0.41≤k≤0.60 and 0.61≤k≤0.80, respectively.26 The Spearman correlation coefficient testing was also used to determine the relationship of PTEN and MET expression levels between primary tumors and corresponding metastases. Cox proportional hazards models were used for univariate and multivariate analysis to estimate and test demographic characteristics, clinical features and biologic parameters for their associations with overall survival. Variables associated with overall survival at univariate analysis were considered for multivariate analysis.
All statistical tests were two sided. Significance levels were set at P≤0.05. All statistical analyses were carried out using the SPSS System V. 19 Software (SPSS Inc., Chicago, IL, USA)
Results
Patient’s Clinicopathological Characteristics
Eighty-three patients with paired primary colorectal tumors and corresponding liver (n=71; 86%), lung (n=6; 7%) or other (n=6; 7%) metastases were enrolled in the present study. Patient’s demographics, clinical and pathological characteristics are listed in Supplementary Table S1. KRAS mutations were not associated with any of the pathological characteristics. On the contrary, a significant association between the PIK3CA mutations and mucinous adenocarcinoma (P<0.0001) and the BRAF mutations and right-sided tumor localization (P=0.049) was observed (Supplementary Table S1). In addition, loss of nuclear expression of PTEN was associated with right-sided tumors (P=0.011) and high histological grade (P=0.024) ,whereas MET overexpression was associated with mucinous adenocarcinoma (P=0.01).
Survival and clinical data were available for 68 patients; none of the 68 patients received therapy before resection of the primary tumor. Fifty-one (75%) patients received all three active cytotoxic drugs (fluorouracil/leucovorin (5-FU/LV), irinotecan and oxaliplatin) and 17 (25%) received a regimen 5-FU/LV with either irinotecan or oxaliplatin. Thirty-four (50%) patients received combination chemotherapy with anti-EGFR monoclonal antibody (cetuximab or panitimumab) and 36 (53%) combination chemotherapy with anti-VEGF monoclonal antibody (bevacizumab). The median time interval between resection of the primary tumor and development of metachronous metastasis was 22 months. Fourteen (28%) patients with synchronous metastases and 14 (78%) with metachronous metastases received oxaliplatin or irinotecan based chemotherapy before biopsy or resection of metastases. At a median follow-up of 71.8 (4.63–136.7) months, 42(62%) patients had died. The median overall survival was 44.8 months (37.9 months for patients with synchronous (n=50) and 79.6 for those with metachronous (n=18) metastases (P=0.013)).
Molecular Characteristics of Primary Colorectal Tumors
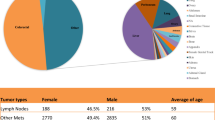
KRAS mutations were detected in 36 (43%), BRAF mutations in three (4%) and PIK3CA mutations in 11 (13%; seven in exon 9 and four in exon 20) out of 83 genotyped primary tumors, respectively (Figure 1). Among KRAS mutations identified, 32 (89%) affected codon 12 and four (11%) codon 13. KRAS and BRAF mutations were mutually exclusive. One BRAF mutant primary tumor harbored the c.1633G>A (p.E545K) PIK3CA mutation. Nine (25%) of the 36 tumors with mutant KRAS and two (4%) of the 47 wild-type KRAS tumors carried PIK3CA mutations (P=0.008, Figure 2e), indicating a significant association between the PIK3CA and KRAS mutations, which seems to be driven by the helical (exon 9) and not the kinase domain (exon 20) mutations (P=0.039 vs P=0.312); among the PIK3CA mutations co-occurring with KRAS mutations, three harbored the c.1624G>A (p.E542K), three the c.1633G>A (p.E545K) and three the c.3140A>G (p.H1047R). Overall, 40 (48%) patients harbored a mutation in any of the KRAS, BRAF and PIK3CA genes.

Proportion of the primary tumors and metastases with KRAS and PIK3CA mutations, high MET gene copy number, overexpression of MET and loss of nuclear and cytoplasmic expression of PTEN and distribution of these molecular alterations from primary tumor to metastases.

Percentage of high MET gene copy number (a, f), overexpression of MET (b, g), loss of cytoplasmic (c, h) and nuclear expression (d, i) of PTEN and PIK3CA mutations (e, j) in KRAS wild-type and KRAS-mutant primary tumors and metastases. P-values indicate the significance of the association between the above-mentioned molecular alterations and KRAS mutations.
Loss of cytoplasmic and nuclear expression of PTEN was detected in 10 (13%) out of 79 and 17 (21%) out of 81 primary tumors, respectively; in addition, loss of both cytoplasmic and nuclear expression of PTEN was detected in only three tumors (P=0.420). All tumors with loss of cytoplasmic expression of PTEN were KRAS wild type (P=0.002, Figure 2c).
MET overexpression and high gene copy number of MET were observed in eight (10%) of 79 and in 21 (31%) of 67 tumors, respectively (Figure 1). None of the eight MET-overexpressed primary tumors had high gene copy numbers of MET while 42% of KRAS wild-type primary tumors had high gene copy numbers of MET (P=0.036; Figure 2a).
Molecular Characteristics of Metastases
KRAS mutations were detected in 31 (37%), BRAF mutations in three (3%) and PIK3CA mutations in 11 (13%; six in exon 9 and five in exon 20) out of 83 metastases, respectively (Figure 1). A trend for an increased incidence of PIK3CA mutations in KRAS-mutant metastases was observed (P=0.091; Figure 2j). Overall, 38 (42%) patients harbored a mutation in any of the KRAS, BRAF and PIK3CA genes.
Loss of cytoplasmic and nuclear expression of PTEN was detected in 13 (18%) of 74 and 13 (16%) of 80 metastases, respectively, while loss of both cytoplasmic and nuclear expression was detected in two metastases. Eleven (37%) KRAS mutant and two (4%) KRAS wild-type metastatic tumors were negative for nuclear expression of PTEN, indicating a significant positive association between the loss of nuclear expression of PTEN and KRAS mutations in metastases (P<0.0001; Figure 2i). Among the eleven KRAS-mutant tumors with loss of nuclear expression of PTEN, five were metachronous and six synchronous metastases.
MET overexpression and high gene copy number of MET was observed in 14 (18%) of 76 and 21 (34%) of 62 metastases, respectively (Figure 1); four (7%) metastases had concurrent overexpression and high gene copy number of MET. None of the metastases with high gene copy number of MET had loss of nuclear expression of PTEN (P=0.012). The incidence of either MET overexpression or high gene copy number of MET was not significantly different between synchronous (22%) and metachronous (6%) metastases (P=0.261) although 10 out of 11 MET-overexpressed metastases were synchronous.
Heterogeneity between Primary Tumors and Corresponding Metastases
KRAS
KRAS mutations were detected by Sanger sequencing in 32 (39%) primary tumors and 22 (27%) related metastases; 10 (12%) patients had a KRAS mutation in their primary but not the metastatic tumor. The wild-type KRAS pairs (51 patients) and the 10 pairs with KRAS discordant results were re-evaluated by PNA Clamp allele-specific real-time PCR assay. Using this assay, 7 out of the 10 discordant paired specimens became concordant; moreover, KRAS mutations could be detected in four additional tumors, which had been characterized as KRAS wild type by Sanger sequencing (Table 1). Finally, five (6%) patients had KRAS mutations exclusively detected in their primary tumors but not in corresponding synchronous metastases (Cohen’s κ=0.875, P<0.001; Figure 1).
BRAF
In all patients, the same classical BRAF c.1799T>A (p.V600E) point mutation was observed both in the primary tumor and the corresponding metastasis.
PIK3CA
PIK3CA mutation analysis by standard cycle sequencing revealed discrepancy between the primary tumors and the corresponding metastases in 12 (15%) patients. Following analysis of these 12 discordant paired specimens by ARMS allele-specific PCR combined with Scorpions probes (designed to detect c.1633G>A (p.E545K), c.1624G>A (p.E542K), c.3140A>G (p.H1047R), c.3140A>T (p.H1047L)), discrepancy was confirmed in six (7%) patients (Cohen’s κ=0.581, P<0.001); in four of them the discrepancy concerned the exon 9 and in two the exon 20 PIK3CA mutation status; however, the PIK3CA mutation status of the two primary tumors showing gain of the c.1634A>G (p.E545G) and c.3139C>T (p.H1047Y) in metastases could not be confirmed by the ARMS-Scorpion assay (Table 2). Among patients with PIK3CA discordant results, four (5%) changed genotype from wild type to mutant (two patients gained exon 9 and two exon 20 mutations) and four (5%) lost their mutations in metastases (Figure 1). Gain of PIK3CA mutations were identified only in synchronous metastases and loss in three synchronous and one metachronous metastases.
PTEN
PTEN cytoplasmic expression was successfully determined in 71 paired specimens. There was no correlation between PTEN cytoplasmic expression in the primary tumors and the corresponding metastases (Spearman’s ρ=0.095; P=0.431). Nine (13%) tumors had lost cytoplasmic expression of PTEN and seven (10%) had gained expression in metastases (Cohen’s κ=0.141; P=0.233).
PTEN nuclear expression was successfully determined in 78 paired specimens. There was no correlation of PTEN nuclear expression between primary tumors and corresponding metastases (Spearman’s ρ=0.078; P=0.495). Eleven (14%) tumors had lost nuclear expression of PTEN and 14 (18%) had gained expression in metastases (Cohen’s κ=0.056; P=0.616).
MET
The expression of MET was successfully determined in 73 paired specimens. There was a positive correlation between MET expression in primary tumors and corresponding metastases (Spearman’s ρ=0.300; P=0.01) while the percentage of agreement was low (Cohen’s κ=0.290; P=0.007). Three (4%) tumors had lost expression of MET and 10 (14%) had gained expression in metastases (Figure 1).
The level of gene copy number alterations of MET was assessed in 59 paired specimens; there was a significant positive correlation of gene copy number of MET between primary tumors and corresponding metastases (Spearman’s ρ=0.516; P<0.0001), while the percentage of concordance was low (Cohen’s κ=0.403; P=0.002). Five (9%) tumors lost the amplification of MET and nine (15%) changed genotype from non-amplified to amplified MET in metastases (Figure 1).
Intra-tumoral Heterogeneity
Using KRAS and PIK3CA mutation status, an exploratory analysis for intra-tumoral heterogeneity was performed in six paired specimens, classified as concordant by the mutant-enriched PCR assays; the mutation status of two different areas either of the primary tumors (three paired specimens) or metastases (three paired specimens) was examined both by Sanger sequencing and the mutant-enriched real-time PCR assay. Intra-tumoral heterogeneity was detected only in primary tumors; two KRAS c.35G>T (p.G12V) mutant tumors also comprised cancer cells harboring the KRAS c.37G>T (p.G13C) mutation or wild-type KRAS, respectively, and one KRAS and PIK3CA wild-type tumor also comprised cells harboring the c.38G>A (p.G13D) and c.3140A>G (p.H1047R) mutations (Table 3).
Association of Molecular and Pathological Tumor Characteristics with Survival
Univariate analysis revealed that the presence of synchronous metastases, the right-sided tumor location, administration of chemotherapy without biological compounds, loss of nuclear expression of PTEN in primary tumors, presence of PIK3CA mutations and overexpression of MET in metastases were significantly associated with shorter overall survival (Supplementary Table S2). The association of MET overexpression with overall survival remained significant even when univariate analysis was limited to only the subgroup of patients with synchronous metastases (24.1(n=10) vs 43.6(n=36) months; P<0.0001). Notably, even when survival was measured from the date of biopsy/resection of metastases, MET overexpression was still associated with shorter survival (17(n=11) vs 37(n=51) months; P<0.0001), whereas PIK3CA mutations (28(n=10) vs 34(n=58) months; P=0.101) did not.
The multiple regression analysis revealed that the right-sided tumor localization and MET overexpression were independently and significantly associated with shorter overall survival (Table 4 and Figure 3).

Kaplan–Meier plot to predict overall survival according to MET immunoexpression status in metastases.
Discussion
In the era of personalized cancer therapy, tailored treatment based on gene alterations and molecular marker expression has become a standard practice. In colorectal cancer, it is documented that KRAS mutations predict unresponsiveness to EGFR-targeted monoclonal antibody therapies; however, about 25% of patients not responding to EGFR-targeted therapy are a wild-type for KRAS, BRAF, PIK3CA and PTEN status. In these ‘quadruple negative’ patients, resistance to EGFR-targeted therapies may be mediated by the activation of parallel pathways such as the MET receptor tyrosine kinase signaling pathway. Moreover, the predictive vs the prognostic value of these molecular biomarkers is still a matter of controversy. Especially in metastatic colorectal cancer the degree of concordance between primary tumors and related metastases regarding the status of all these promising biomarkers may be of great importance for decision making and prediction of the efficacy of targeted therapies as metastasis mirrors the actual disease status.
To our knowledge, the current study is the first one reporting on the correlation of mutational status of KRAS, BRAF and PIK3CA with cytoplasmic and nuclear expression of PTEN, as well as the expression and the gene copy number of MET in primary colorectal tumors and corresponding metastases; moreover, this genetic characterization of primary and metastatic tumors was correlated with patients’ clinical outcome.
Our findings demonstrate a high level of concordance between KRAS and BRAF mutation status in primary tumors and related metastases and point to a certain degree of intra-tumoral heterogeneity in primary tumors, which is in agreement with previous studies.11, 12 Furthermore, our data emphasize the importance of using sensitive molecular methods to ensure efficient mutation detection especially in metastases as the failure to detect a mutation using the Sanger sequencing may merely indicate that the mutation is present in <20% of the analyzed DNA template molecules. Nevertheless, different KRAS genotypes were observed in 6% of the paired specimens, indicating that in order to restrict EGFR-targeted therapy only to patients with wild-type KRAS tumors it could be more appropriate to perform KRAS genotyping in metastases rather than the primary tumors when biological material is available. It is obvious that the clinical utility of this remains to be proven in prospective well-designed clinical studies.
PIK3CA mutation status was found to be discordant in 10% of the analyzed paired specimens; a previous study in colorectal cancer reported a discordance rate of 5%, whereas in breast cancer the PIK3CA mutation status frequently changes during disease progression.12, 27, 28 One important finding in the current study is the significant association of KRAS and PIK3CA mutations in primary tumors (P=0.008), which seems to be driven by the helical (exon 9) mutations (P=0.039) as kinase domain (exon 20) mutations alone do not associate with KRAS mutations (P=0.312). Conversely, a similar association could not be observed in metastases, despite the existence of a statistical trend (P=0.092); this is in line with the observed discrepancy of PIK3CA mutation status in the paired specimens and could possibly have an impact in determining the optimal algorithm for mutation screening in metastatic colorectal cancer. However, the small number of patients harboring PIK3CA mutations in the present study precludes any definitive conclusion and these results remain hypothesis-generating. Recently, De Roock et al29 studying a large cohort of patients with metastatic colorectal cancer arrived at the same conclusion concerning the association between KRAS and exon 9 PIK3CA mutations; in addition, the authors proposed that in KRAS wild-type tumors, only the PIK3CA exon 20 mutations have a negative effect on the efficacy of EGFR-targeted therapy.
PTEN expression and localization were heterogeneous between paired specimens. The detection of PTEN by immunohistochemistry instead of other molecular analyses was chosen because it covers both genetic and epigenetic mechanisms underlying loss of PTEN function.30 Nuclear and cytoplasmic expression of PTEN were evaluated by using two different antibodies displaying predominantly nuclear and cytoplasmic staining, respectively.31, 32, 33, 34 Loss of PTEN cytoplasmic expression was observed only in primary tumors with KRAS wild-type status. Previous studies have shown that RAS mutations typically arise in PTEN wild-type genetic background, indicating that RAS activation and loss of PTEN probably serve the same function during tumorigenesis.35, 36, 37 On the other hand, although RAS is a potent oncogene, its tumorigenicity depends on the cellular context and cooperative events and previous reports have shown that expression of PTEN is suppressed by the oncogenic RAS via the RAF/ERK/MEK signaling pathway giving rise to cells with greater malignant potential.38, 39, 40, 41, 42 The localization of PTEN has not been assessed in these studies although accumulating genetic, pathologic and biochemical evidence suggests that the localization of PTEN either in the nucleus or cytoplasm affects the proliferation of tumor cells.43, 44, 45, 46, 47 In the current study, it was found that loss of PTEN nuclear expression in metastases was significantly associated with KRAS mutations; this finding is in accordance with a mechanism of suppression of PTEN nuclear translocation through KRAS activation. Although the association of KRAS mutations with the loss of PTEN nuclear expression seems to be contradictory with the observed loss of PTEN cytoplasmic expression only in tumors with KRAS wild type, these findings could be explained if we hypothesize that the subcellular localization of PTEN is affected by different genetic or epigenetic alterations that, probably, may co-occur or not with KRAS mutations. However, our results should be considered with caution as a common drawback of all studies evaluating expression of PTEN by immunohistochemistry is the lack of a robust universally-acceptable PTEN immunohistochemistry assay and because the differences observed between primary tumors and related metastases could be the result of previous medical treatments.31, 48
Previous studies have shown that loss of PTEN nuclear expression and BRAF mutations were associated with poor clinical outcome while the prognostic significance of PIK3CA and KRAS mutations in colorectal cancer remains elusive.24, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58 In our series, loss of PTEN nuclear expression in primary tumors and the PIK3CA mutations in metastases were associated with shorter overall survival (Supplementary Table S2); however, neither PTEN expression nor PIK3CA mutations were emerged as independent prognostic factors in multivariate analysis (Table 4). Activation of RAS/PI3K signaling network by mutation of at least one of the BRAF, KRAS and PI3KCA was associated with shorter survival, as previously reported by Barault et al (data not shown).54
Aberrant activation of the MET signaling pathway has been implicated in the development and progression of colorectal cancer.7, 59, 60, 61, 62 Our findings demonstrate that a high MET gene copy number in primary tumors was correlated with KRAS wild type while in metastases it was inversely correlated with the loss of nuclear PTEN expression. These data, combined with the positive association of KRAS mutations and loss of PTEN nuclear expression in metastases, seem to indicate a distinct role of MET signaling in KRAS wild-type metastases. Nevertheless, the most frequent cause of constitutive activation of MET in human tumors is increased protein expression in the absence of gene amplification;63 accordingly, in the present study the expression of MET as measured by immunohistochemistry was not found to correlate with the amplification of MET as already has been described.64
However, the data reported on MET gene copy number are in disagreement with previous studies showing that MET amplification is a rare event in primary colorectal cancer tumors.65, 66 This discrepancy may be due to methodological differences as we used a quantitative RT-PCR assay instead of FISH and thus the reported increase in MET gene copy number could be due to a polysomy of chromosome 7 and not locus specific amplification of MET. Moreover, the choice of TOP3A as a reference gene in our experiments, which resides on chromosome 17 whose loss has been associated with transition to carcinoma, may be another reason for this discrepancy; however this possibility was investigated using KRAS as a reference gene and similar results were obtained (unpublished results).
In the current study, the MET gene copy number was not significantly associated with overall survival, whereas MET overexpression in metastases emerged as an independent factor associated with shorter overall survival. Several previous studies have shown that MET polysomy is correlated with high levels of MET mRNA and that high levels of MET mRNA were associated with tumor invasion; however, no prognostic significance has been reported for either MET polysomy or MET amplification.25, 67 Furthermore, previous studies have provided evidence for the prognostic value of MET protein expression in the early stages of carcinoma progression.68, 69 In this study the majority of patients had metastatic disease and they all received the appropriate chemotherapy although their access to EGFR- or VEGF-targeted therapy was not always based on molecular biomarker selection; therefore, the data reported herein highlight the value of MET expression in metastases as a biomarker that might be used to select advanced colorectal cancer patients who could benefit from targeted therapies.
In conclusion, the current study demonstrates (1) a certain degree of discordance between primary colorectal cancer tumors and related metastases regarding all examined biomarkers except BRAF mutations; (2) an association between KRAS and the helical PIK3CA mutations; (3) an association between KRAS mutations and loss of PTEN nuclear expression and (4) a possible prognostic value of MET overexpression in metastatic disease. Overall, it appears that metastatic lesions are the appropriate tissues to analyze in order to determine targeted therapies in metastatic colorectal cancer and the activation RAS/PI3K and MET signaling pathways defines distinct subsets of metastatic colorectal cancer patients.
References
Ross JS, Torres-Mora J, Wagle N et al Biomarker-based prediction of response to therapy for colorectal cancer: current perspective. Am J Clin Pathol 2010;134:478–490.
Segal NH, Saltz LB . Evolving treatment of advanced colon cancer. Annu Rev Med 2009;60:207–219.
De Roock W, De Vriendt V, Normanno N et al KRAS, BRAF, PIK3CA and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 2011;12:594–603.
Prenen H, De Schutter J, Jacobs B et al PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 2009;15:3184–3188.
Sartore-Bianchi A, Martini M, Molinari F et al PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009;69:1851–1857.
Souglakos J, Philips J, Wang R et al Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 2009;101:465–472.
Liska D, Chen CT, Bachleitner-Hofmann T et al HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res 2010;17:472–482.
Krumbach R, Schuler J, Hofmann M et al Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer 2011;47:1231–1243.
Gray J . Cancer: genomics of metastasis. Nature 2010;464:989–990.
Jones S, Chen WD, Parmigiani G et al Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA 2008;105:4283–4288.
Baas JM, Krens LL, Guchelaar HJ et al Concordance of predictive markers for EGFR inhibitors in primary tumors and metastases in colorectal cancer: a review. Oncologist 2011;16:1239–1249.
Baldus SE, Schaefer KL, Engers R et al Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 2010;16:790–799.
Knijn N, Mekenkamp LJ, Klomp M et al KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer 2011;104:1020–1026.
Sayagues JM, Abad Mdel M, Melchor HB et al Intratumoural cytogenetic heterogeneity of sporadic colorectal carcinomas suggests several pathways to liver metastasis. J Pathol 2010;221:308–319.
Sleeman J, Steeg PS . Cancer metastasis as a therapeutic target. Eur J Cancer 2010;46:1177–1180.
Klein CA . Parallel progression of primary tumours and metastases. Nat Rev Cancer 2009;9:302–312.
Ju HX, An B, Okamoto Y et al Distinct profiles of epigenetic evolution between colorectal cancers with and without metastasis. Am J Pathol 2011;178:1835–1846.
Watanabe T, Kobunai T, Yamamoto Y et al Differential gene expression signatures between colorectal cancers with and without KRAS mutations: crosstalk between the KRAS pathway and other signalling pathways. Eur J Cancer 2011;47:1946–1954.
Molinari F, Martin V, Saletti P et al Differing deregulation of EGFR and downstream proteins in primary colorectal cancer and related metastatic sites may be clinically relevant. Br J Cancer 2009;100:1087–1094.
Kalikaki A, Koutsopoulos A, Trypaki M et al Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer 2008;99:923–929.
Board RE, Wardley AM, Dixon JM et al Detection of PIK3CA mutations in circulating free DNA in patients with breast cancer. Breast Cancer Res Treat 2010;120:461–467.
Smolen GA, Sordella R, Muir B et al Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci USA 2006;103:2316–2321.
Loupakis F, Pollina L, Stasi I et al PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 2009;27:2622–2629.
Jang KS, Song YS, Jang SH et al Clinicopathological significance of nuclear PTEN expression in colorectal adenocarcinoma. Histopathology 2010;56:229–239.
Takeuchi H, Bilchik A, Saha S et al c-MET expression level in primary colon cancer: a predictor of tumor invasion and lymph node metastases. Clin Cancer Res 2003;9:1480–1488.
Landis JR, Koch GG . The measurement of observer agreement for categorical data. Biometrics 1977;33:159–174.
Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K et al PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther 2011;10:1093–1101.
Dupont Jensen J, Laenkholm AV, Knoop A et al PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin Cancer Res 2010;17:667–677.
De Roock W, Claes B, Bernasconi D et al Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753–762.
Salmena L, Carracedo A, Pandolfi PP . Tenets of PTEN tumor suppression. Cell 2008;133:403–414.
Pallares J, Bussaglia E, Martinez-Guitarte JL et al Immunohistochemical analysis of PTEN in endometrial carcinoma: a tissue microarray study with a comparison of four commercial antibodies in correlation with molecular abnormalities. Mod Pathol 2005;18:719–727.
Tian T, Nan KJ, Wang SH et al PTEN regulates angiogenesis and VEGF expression through phosphatase-dependent and -independent mechanisms in HepG2 cells. Carcinogenesis 2010;31:1211–1219.
Gazzola A, Bertuzzi C, Agostinelli C et al Physiological PTEN expression in peripheral T-cell lymphoma not otherwise specified. Haematologica 2009;94:1036–1037.
Torres J, Navarro S, Rogla I et al Heterogeneous lack of expression of the tumour suppressor PTEN protein in human neoplastic tissues. Eur J Cancer 2001;37:114–121.
Mao JH, To MD, Perez-Losada J et al Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev 2004;18:1800–1805.
Ikeda T, Yoshinaga K, Suzuki A et al Anticorresponding mutations of the KRAS and PTEN genes in human endometrial cancer. Oncol Rep 2000;7:567–570.
Tsao H, Zhang X, Fowlkes K et al Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res 2000;60:1800–1804.
Bahk YY, Cho IH, Kim TS . A cross-talk between oncogenic Ras and tumor suppressor PTEN through FAK Tyr861 phosphorylation in NIH/3T3 mouse embryonic fibroblasts. Biochem Biophys Res Commun 2008;377:1199–1204.
Vasudevan KM, Burikhanov R, Goswami A et al Suppression of PTEN expression is essential for antiapoptosis and cellular transformation by oncogenic Ras. Cancer Res 2007;67:10343–10350.
Hill R, Calvopina JH, Kim C et al PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res 2010;70:7114–7124.
Nogueira C, Kim KH, Sung H et al Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010;29:6222–6232.
Iwanaga K, Yang Y, Raso MG et al Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res 2008;68:1119–1127.
Shen WH, Balajee AS, Wang J et al Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007;128:157–170.
Song MS, Carracedo A, Salmena L et al Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011;144:187–199.
Wrighton KH . Cancer biology: Role of nuclear PTEN revealed. Nat Rev Mol Cell Biol 2011;12:134.
Wrighton KH . Tumour suppressors: Role of nuclear PTEN revealed. Nat Rev Cancer 2011;11:154.
Chung JH, Eng C . Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res 2005;65:8096–8100.
Sangale Z, Prass C, Carlson A et al A robust immunohistochemical assay for detecting PTEN expression in human tumors. Appl Immunohistochem Mol Morphol 2011;19:173–183.
Ogino S, Nosho K, Kirkner GJ et al CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009;58:90–96.
Ogino S, Shima K, Meyerhardt JA et al Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res 2009;18:890–900.
Samowitz WS, Sweeney C, Herrick J et al Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65:6063–6069.
Ogino S, Nosho K, Kirkner GJ et al PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol 2009;27:1477–1484.
He Y, Van’t Veer LJ, Mikolajewska-Hanclich I et al PIK3CA mutations predict local recurrences in rectal cancer patients. Clin Cancer Res 2009;15:6956–6962.
Barault L, Veyrie N, Jooste V et al Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer 2008;122:2255–2259.
Hsu CP, Kao TY, Chang WL et al Clinical significance of tumor suppressor PTEN in colorectal carcinoma. Eur J Surg Oncol 2011;37:140–147.
Barault L, Charon-Barra C, Jooste V et al Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res 2008;68:8541–8546.
Samowitz WS, Curtin K, Schaffer D et al Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev 2000;9:1193–1197.
Liao X, Morikawa T, Lochhead P et al Prognostic role of PIK3CA mutation in colorectal cancer: Cohort Study and literature review. Clin Cancer Res 2012;18:2257–2268.
Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG . Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol 2009;10:709–717.
Boon EM, Kovarikova M, Derksen PW et al MET signalling in primary colon epithelial cells leads to increased transformation irrespective of aberrant Wnt signalling. Br J Cancer 2005;92:1078–1083.
Long IS, Han K, Li M et al Met receptor overexpression and oncogenic Ki-ras mutation cooperate to enhance tumorigenicity of colon cancer cells in vivo. Mol Cancer Res 2003;1:393–401.
Rasola A, Fassetta M, De Bacco F et al A positive feedback loop between hepatocyte growth factor receptor and beta-catenin sustains colorectal cancer cell invasive growth. Oncogene 2007;26:1078–1087.
Comoglio PM, Giordano S, Trusolino L . Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov 2008;7:504–516.
Janjigian YY, Tang LH, Coit DG et al MET expression and amplification in patients with localized gastric cancer. Cancer Epidemiol Biomarkers Prev 2011;20:1021–1027.
Galimi F, Torti D, Sassi F et al Genetic and expression analysis of MET, MACC1, and HGF in metastatic colorectal cancer: response to met inhibition in patient xenografts and pathologic correlations. Clin Cancer Res 2011;17:3146–3156.
Cappuzzo F, Varella-Garcia M, Finocchiaro G et al Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br J Cancer 2008;99:83–89.
Zeng ZS, Weiser MR, Kuntz E et al c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett 2008;265:258–269.
Ginty F, Adak S, Can A et al The relative distribution of membranous and cytoplasmic met is a prognostic indicator in stage I and II colon cancer. Clin Cancer Res 2008;14:3814–3822.
Resnick MB, Routhier J, Konkin T et al Epidermal growth factor receptor, c-MET, beta-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Cancer Res 2004;10:3069–3075.
Acknowledgements
This work was supported by a research grant from the Cretan Association for Biomedical Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article
Cite this article
Voutsina, A., Tzardi, M., Kalikaki, A. et al. Combined analysis of KRAS and PIK3CA mutations, MET and PTEN expression in primary tumors and corresponding metastases in colorectal cancer. Mod Pathol 26, 302–313 (2013). https://doi.org/10.1038/modpathol.2012.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.150
Keywords
This article is cited by
-
Relationship between KRAS mutations and dual time point 18F-FDG PET/CT imaging in colorectal liver metastases
Abdominal Radiology (2019)
-
Prognostic significance of CEACAM5mRNA-positive circulating tumor cells in patients with metastatic colorectal cancer
Cancer Chemotherapy and Pharmacology (2018)
-
Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges
Nature Reviews Clinical Oncology (2017)
-
High sensitivity isoelectric focusing to establish a signaling biomarker for the diagnosis of human colorectal cancer
BMC Cancer (2016)
-
c-MET immunostaining in colorectal carcinoma is associated with local disease recurrence
BMC Cancer (2015)

{kind=link}
{kind=link}